-
Синдром Дауна
-
Р
ис.
15 –Зовнішній вигляд дітей з синдромом
Дауна
-
Порушення
нормального функціонування систем
організму також породжує деякі зовнішні
особливості. Зокрема, у хворих нерідко
спостерігається гіперрухливість
суглобів і недостатній м'язовий тонус.
Дані стану спостерігаються приблизно
у
80% випадків. Через збільшений
язик
(макроглосія) і своєрідної
будови піднебіння,
рот хворого завжди відкритий. М'язи
особи не здатні утримувати губи в
закритому положенні. У 65% випадків
виявляються аномалії зубів.
-
Характерні риси
розумового розвитку:
-
• Помітне
відставання у
розвитку. Навіть при своєчасному
комплексному лікуванні затримки в
розвитку будуть все помітніше з віком.
Розвиток розумових здібностей, в
більшості випадків, залишається на
рівні семирічної дитини. У рідкісних
випадках інтелект досягає більш високого
рівня.
-
• Невеликий
словниковий запас. Крім того, що пацієнти
з даною аномалією невиразно говорять,
відзначається досить мізерний мовний
набір.
-
• Відсутність
здібностей до абстрактного мислення.
Таким дітям легше розуміти і аналізувати
тільки те, що вони бачать безпосередньо
перед очима, уявити якусь ситуацію або
пофантазувати для них вже проблематично.
-
• Низька
концентрація уваги. Дитині важко
сфокусуватися на будь-яку
задачу,
досить швидко вони починають відволікатися.
-
Синдром
Дауна - це практично єдина хромосомна
аномалія, коли діагноз можна поставити
клінічно, тобто, орієнтуючись тільки
на зовнішні ознаки. Однак в будь-якому
випадку необхідно буде провести
каріотипування, щоб визначити форму
синдрому.
-
Окрім наявності
самого синдрому Дауна у хворих практично
завжди відзначається ряд інших проблем
зі здоров'ям. Носії цієї хромосомної
аномалії дуже часто стикаються з недугами
кардіологічного характеру. Вроджені
вади серця відзначаються приблизно в
40% випадків. Порушення біологічних
процесів тягне за собою також ранній
розвиток катаракти, з яким стикаються
дві третини хворих старше восьми років.
При синдромі Дауна значно збільшується
ризик розвитку хвороби Альцгеймера і
появи гострого мієлоїдного лейкозу. У
пацієнтів з даною патологією часто є
відхилення в роботі шлунково-кишкового
тракту. Рекомендуються регулярні
обстеження у гастроентеролога і
кардіолога.
-
Простудні
захворювання, ГРВІ та пневмонії -
регулярні супутники хворого синдромом
Дауна. Це пояснюється ослабленою імунною
системою організму.
-
Поява зайвої
хромосоми в організмі викликає також
порушення обміну речовин, що призводить
до несправного функціонування внутрішніх
органів (захворювання щитовидної залози,
порушення зору, слуху і т.д).
-
Треба зауважити,
що результати деяких досліджень вказують
на те, що люди, які є носіями цієї
хромосомної аномалії, мають відносно
невисоку частоту виникнення злоякісних
пухлин в організмі. Однак накопичена
на сьогоднішній день інформація щодо
даного медичного питання не дає змоги
з упевненістю говорити про причини
такого взаємозв'язку.
-
Лікування
-
Генетична
природа захворювання зводить шанси на
успішну боротьбу з ним до мінімуму.
Однак імовірність того, що все-таки
будуть розроблені методики, що дозволяють
проводити успішне лікування синдрому
Дауна. Дослідження
вчених, проведені в останні роки,
показали, що можна зробити своєрідне
"виключення" зайвої копії 21
хромосоми. Щоб здійснити дану дію,
передбачається використовувати особливу
поведінку
X-хромосоми, яка відповідає за приналежність
людини до жіночої статі. Чоловічим
набором хромосом, як відомо, є XY, а жіночим
- набір XX. Вченими було встановлено, що
в організмі жінки у одній з хромосом
спостерігається інактивований
стан, а її дані не використовуються в
процесі синтезу білкових структур. По
суті, вона не є учасником системи
біохімічних реакцій в організмі.
Відбувається це внаслідок вироблення
особливих речовин під керуванням
специфічної
ділянки ДНК. Ці речовини покривають
поверхню хромосоми і, тим самим, блокують
доступ до неї.
-
Медики
зробили спробу скористатися цим
механізмом, з метою зробити неактивною
третю копію 21 хромосоми. Дані досліджень
показують, що саме її активність в
процесі виробництва білкових з'єднань
ініціює
появу різноманітних відхилень від норм
розвитку. Для реалізації задумки
необхідно було зробити зайвий екземпляр
21 хромосоми схожим на X-хромосому. Для
цього вчені застосували кілька
модифікованих
ділянок
генетичного матеріалу, що викликає
інактивацію. Ця ділянка була поставлена
замість одного з «сміттєвих» фрагментів
(частина ДНК, яка відповідає за синтез
білків) в зайву хромосому. Через деякий
час, оброблена подібним чином копія 21
хромосоми припинила проявляти активність.
Дослідники висунули припущення, що це
дозволить компенсувати наявність
зайвого генетичного матеріалу.
-
Необхідно
зауважити, що досліди проводилися тільки
на культурі стовбурових клітин, а
значить, на сьогоднішній день неможна
говорити про будь-яку
ефективність
методу. Крім того, допомога від цього
підходу можуть отримати тільки ті люди
з синдромом Дауна, у яких він обумовлений
трисомією.
Транслокаційну
форму хвороби подібним чином вилікувати
неможливо: довелося б «вимикати» цілу
хромосому, а це однозначно принесе
більше шкоди, ніж користі. Однак, з огляду
на стрімкий прогрес в сфері генетики
та пов'язаних з нею досліджень, можна
сказати, що метод має певні перспективи.
-
Соціалізація
і інтеграція в суспільство
-
Дітей
з синдромом Дауна іноді називають
«сонячними». Пояснюється це тим, що вони
дуже доброзичливі і позитивні, не
дивлячись на можливі перепади настрою
і періодичні спалахи агресії. Завдяки
стрімкому розвитку медицини і педагогіки
у малюків з синдромом Дауна тепер
набагато більше перспектив у отриманні
освіти, ніж це було на початку XX сторіччя.
В ті часи тривалість життя людей з даною
патологією становила в середньому 20-25
років. Вплив цього фактора не дозволяв
їм приділяти достатню кількість часу
освоєння тих чи інших дисциплін. Останні
статистичні дослідження на питання
«скільки живуть люди з синдромом Дауна?»
Відповідають, що планка тривалості
життя піднялася до 50 років.
-
Р
ис.
15 –Зовнішній вигляд дітей з синдромом
Дауна
- Синдром Едвардса
-
Синдром Патау
-
Трисомія
13 або синдром Патау - мутація, патологія,
генне
захворювання, при якому в клітинах
з'являється додаткова тринадцята
хромосома. Стоїть в одному ряду з такими
захворюваннями, як синдром Дауна або
Едвардса. Однак зустрічається набагато
рідше їх і проявляється цілим набором
зовнішніх каліцтв і внутрішніх патологій.
У медицині можна зустріти інші назви
хвороби: трисомія D, синдром Патау-Смітта.
Про причини цієї мутації генетики можуть
тільки здогадуватися.
-
Причини
-
Основні причини
синдрому Патау, виявлені на сьогоднішній
день:
-
• помилки у
формуванні сперматозоїдів і яйцеклітин,
що беруть участь у зачатті дитини, тобто
абсолютно випадковий фактор;
-
• вік матері після
45 років;
-
• несприятлива
екологія, особливо при радіаційному
зараженні;
-
• шлюби між
родичами;
-
• спадковість -
рідкісна причина, так як генетичний
матеріал несе в собі здорова людина, що
не має ознак трисомії 13.
-
Через
це в хромосомному наборі дитини можуть
виникнути патології. Враховуючи цей
список причин, молоді матусі повинні
уникати цих чинників, наскільки це
можливо. А при попаданні в групу ризику
пройти всі необхідні дослідження для
виявлення захворювання. Симптоматика
зумовлена тим, що в основі синдрому
Патау лежить наявність додаткової
хромосоми 13, якої немає у звичайних
людей.
-
Симптоми і ознаки
-
Існують
різні ознаки синдрому Патау, які не
сплутаєш ні з якою іншою хворобою. Так
як це геномна мутація, її симптоми
яскраво виражаються навіть у зовнішності.
Вроджені вади нерідко призводять до
летального результату дітей ще в
дитинстві. Так як в наявності зовнішні
каліцтва, батьки часто відмовляються
від таких діток.
-
Явні УЗД-ознаки
синдрому Патау:
-
• зовнішні каліцтва;
-
• уповільнений
розвиток плода;
-
• тахікардія (в
70% випадків);
-
• багатоводдя (в
50% випадків) - ця ознака дозволяє виявити
синдром Патау на УЗД з урахуванням інших
діагностик;
-
• мегацістіс
(збільшений в розмірах сечовий міхур);
-
• голопрозенцефалія
- нерозділення
мозку на дві півкулі;
-
• омфалоцеле -
ембріональна пуповинна грижа.
-
Зовнішні
симптоми
(рис.18):
-
Р
ис.
18 – Зовнішні симптоми дитини з синдромом
Патау
-
• помірна
мікроцефалія - зменшені розміри черепа
і головного мозку;
-
• низький
лоб, найчастіше скошений;
-
• вузькі
очні щілини на невеликій відстані один
від одного;
-
• мікроофтальмія
- недорозвинення очей;
-
• колобома
- відсутність очної оболонки;
-
• запале
перенісся;
-
• помутніння
рогівки;
-
• деформовані
вушні раковини;
-
• ущелини
піднебіння;
-
• полідактилія
- більша кількість пальців на руках і
ногах, ніж у інших людей
(рис.19);
-
• вигин
кистей;
-
• коротка
шия.
-
• помірна
мікроцефалія - зменшені розміри черепа
і головного мозку;
-
• Порушення
функціонування відділів ЦНС;
-
• вади розвитку
серця (у 80%): дефекти перегородок
(міжшлуночкової і міжпередсердної),
транспозиції судин;
-
• додаткова
селезінка;
-
• фіброкістозні
зміни в підшлунковій залозі;
-
• ембріональна
пупкова грижа;
-
• збільшені нирки;
-
• дольковість
нирок;
-
• наявність в
кірковому шарі нирок кісти;
-
• патології
статевих органів: їх гіпоплазія, у
хлопчиків крипторхізм - неопущення
яєчок, у дівчаток - дворога матка;
-
• затримка
розумового розвитку;
-
• відсутність
задньої стінки сечівника.
-
Основні, типові
ознаки захворювання виявляються при
УЗД вже на 12 тижні вагітності, а потім
підтверджуються всілякими діагностичними
дослідженнями. Якщо вони не проводилися
пренатально, клінічна картина синдрому
Патау після народження малюка досить
яскраво ілюструє саме цей діагноз. Але
для виключення помилки проводяться
аналізи на каріотип. У генетиці розрізняють
кілька типів патології.
-
Класифікація
-
Розрізняють два
цитогенетичних виду синдрому Патау:
-
• проста трисомія;
-
• робертсонівські
транслокация.
-
Набагато рідше
діагностуються інші цитогенетичні
різновиди захворювання:
-
• мозаїцизм
- наявність в тканинах людини генетично
різних клітин;
-
• ізохромосома;
-
• неробертсонівська
транслокація.
-
Клінічна
картина простих трисомних
і транслокаційний
форм не відрізняється. Всі клітини
дитини мають характерний каріотип
синдрому Патау - 47, XX 13+ або 47, XY 13+.
-
Поряд з іншими
схожими захворюваннями, синдром Патау
- це генетична мутація, природу якої
ученим ще тільки належить з'ясувати.
Однак від тих же синдромів Дауна,
Шерешевського-Тернера, Едвардса, котячого
крику, Клайнфельтера дану патологію
відрізняють такі особливості:
-
• згідно з останніми
даними, частота народження синдрому
Патау - один випадок на 10 000;
-
• між цим показником
віком матері простежується нечітка,
але все-таки залежність, вона менш
сувора, ніж при народженні дітей з
синдромом Дауна;
-
• малюки народжуються
з пренатальної гіпоплазією (недорозвиненістю
тканин і органів), яка не пояснюється
недоношенностью (середній термін таких
вагітностей становить зазвичай близько
38,5 тижнів);
-
• зовнішні
і внутрішні каліцтва не дозволяють
таким дітям вести нормальний спосіб
життя
(рис.20).
-
Незважаючи на те,
що сьогодні ведеться активна пропаганда
проти абортів, а синдром Патау - хвороба
дуже рідкісна, якщо вона була виявлена
на ранніх термінах вагітності, лікарі
однозначно порадять її екстрене
переривання. Це позбавить і батьків, і
ще не народженого малюка від безлічі
мук.
-
Діагностика
-
Важливу
роль відіграє діагностика синдрому
Патау, так як вона дозволяє відрізнити
захворювання від подібних генних мутацій
(синдрому Меккеля і Мора, трігоноцефалії
Опітц), які за деякими ознаками збігаються
з трисомєю 13. Вирішальним фактором є
цитогенетичне дослідження хромосом.
Воно необхідне для прогнозу життя та
здоров'я дітей, дозволяє своєчасно
перервати вагітність при підтвердженні
діагнозу.
-
I
етап
-
1. УЗД-дослідження.
-
2. Визначення
біохімічних і фізичних маркерів бета-ХГЧ
(гормону хоріона), РАРР-А (білка плаценти)
і ін.
-
3. Розрахунок шансів
на народження малюка з синдромом Патау.
-
II етап
-
На даному етапі
діагностика проводиться для тих вагітних,
хто потрапив в групу ризику за підсумками
досліджень I етапу.
-
1. 8-12 тижнів: береться
біопсія хоріона;
-
2.
14-18 тижнів: проводиться амніоцентез -
прокол амніотичної оболонки, щоб
дослідити
навколоплідні води;
-
3. 20 тижнів:
кордоцентез - дослідження пуповинної
крові.
-
У
матеріалах, отриманих в ході цих заходів,
генетики шукають патологію методом
КФ-ПЛР або за допомогою каріотипування,
диференційно фарбуючи хромосоми. Якщо
діагностика при вагітності не проводилася,
хромосомну аномалію виявляють на
підставі клінічної картини. Для точної
постановки діагнозу обов'язковий
генетичний аналіз для визначення
каріотипу новонародженого.
-
Лікування
-
Профілактика
-
Трисомія
13 або синдром Патау - мутація, патологія,
генне
захворювання, при якому в клітинах
з'являється додаткова тринадцята
хромосома. Стоїть в одному ряду з такими
захворюваннями, як синдром Дауна або
Едвардса. Однак зустрічається набагато
рідше їх і проявляється цілим набором
зовнішніх каліцтв і внутрішніх патологій.
У медицині можна зустріти інші назви
хвороби: трисомія D, синдром Патау-Смітта.
Про причини цієї мутації генетики можуть
тільки здогадуватися.
-
Синдром Шерешевського –Тернера
-
Синдром Клайнфельтера
ТЕМА 4. ЗАГАЛЬНА ХАРАКТЕРИСТИКА ХРОМОСОМНИХ ХВОРОБ. КЛІНІКА ОСНОВНИХ ФОРМ ХРОМОСОМНИХ ХВОРОБ.
Хромосомні хвороби – велика група природжених спадкових хвороб, які клінічно характеризуються множинними вадами розвитку, в основі яких лежать числові або структурні зміни хромосом.
Найбільш повні дані про частоту і розповсюдження хромосомних хвороб можна отримати на основі цитогенетичних досліджень спонтанних абортів, мертвонароджених і новонароджених з природженими вадами розвитку.
Хромосомні аберації:
Серед новонароджених зустрічаються приблизно у 1% дітей
Серед дітей, які народилися із затримкою психомоторного розвитку і які мають вади розвитку – від 10 до 30%.
Більш як 50% спонтанних абортів у 1 триместрі вагітності пов’язані з хромосомними абераціями.
У хворих з порушенням статевої диференціації від 20 до 50%.
У хворих з первинною і вторинною аменореєю від 10 до 50%.
Структурні хромосомні перебудови у 5% подружніх пар є причиною звичного невиношування вагітності.
У людини знайдено всі форми хромосомних і геномних мутацій. Більшість хромосомних хвороб виникає спорадично у результаті геномної чи хромосомної мутації в гаметах здорових батьків або у разі перших поділів зиготи. Хромосомні зміни в гаметах призводять до розвитку так званих повних або регулярних форм порушення каріотипу, а відповідні зміни хромосом на ранніх стадіях розвитку зиготи є причиною виникнення соматичної мозаїчності, або мозаїчних організмів (наявність в організмі двох або більше клітинних ліній з різним числом хромосом). Зигота, яка розвивається, може бути сформована двома або більше клітинними лініями, співвідношення яких у мозаїчному організмі залежить від стадії, на якій відбулося порушення, а також від життєздатності і темпів проліферації утворюваних клітинних ліній.
В основу класифікації хромосомних хвороб покладено тип хромосомної аномалії і характер дисбалансу хромосомного матеріалу відповідного каріотипу.
Виходячи з цих принципів хромосомні аномалії каріотипу поділяються на 3 групи:
кількісні порушення за окремими хромосомами (анеуплодії),
порушення кратності повного гаплоїдного набору хромосом (поліплоїдії),
структурні перебудови хромосом.
Перші дві групи відносяться до геномних мутацій, а третя група – до хромосомних.
Поліплоїдія виникає в результаті порушення нормального мітотичного циклу, подвоєння хромосом не супроводжується поділом ядра і клітини. Прикладом є триплоїдія (69, ХХХ) і тетраплоїдія (92, ХХХХ), які частіше зустрічаються в матеріалі спонтанних абортів або у плоді і мертвонароджених, а інколи і в новонароджених, в яких тривалість життя з такими аномаліями складає, як правило всього декілька днів.
Анеуплоїдія (збільшення або зменшення однієї або декількох хромосом), виникає в результаті нерозходження хромосом в мейотичних поділах або в мітозі. Термін «нерозходження» означає відсутність розладнання хромосом (в мейозі) або хроматид (в мітозі) в анафазі. В результаті нерозходження виникають гамети з аномальним набором хромосом.
Структурні зміни хромосом у людини зустрічаються рідше, ніж числові аберації. Структурні перебудови можуть бути хромосомними і хроматидними, супроводжуватись змінами кількості генетичного матеріалу (делеції і дуплікації), або лише зводяться до його переміщення (інверсії, транслокації). В перебудові може брати участь одна або більше хромосом з декількома розривами і з’єднаннями.
Будь-яка хромосома каріотипу людини може брати участь в кількісних чи структурних змінах. Виходячи з цього, можна спостерігати велику різноманітність описаних хромосомних форм. Практична цитогенетика постійно стикається з виявленням нових хромосомних аномалій при дослідженні різноманітних клітин і тканин в різні періоди розвитку людини.
Характер і важкість клінічного прояву хромосомних хвороб залежить від виду аномалії і ступеня втягнення хромосом.
Діагностика хромосомних синдромів на основі клінічних характеристик не завжди можлива з наступних причин:
відсутня патогномонічна для даного синдрому ознака;
перекривання ознак різних хромосомних синдромів;
варіабельність фенотипічних проявів одного і того ж синдрому у різних індивідуумів;
вади розвитку, які спостерігаються при хромосомних синдромах можуть зустрічатися і при нормальному каріотипі.
Клініко-цитогенетичне вивчення хромосомних аномалій дозволяє визначити ряд ознак, які в різних поєднаннях і з різним ступенем вираженості зустрічаються при всіх хромосомних синдромах.
При вивченні кореляції фенотипу з каріотипом було зроблено важливе заключення про те, що чим більша кількість хромосомного матеріалу втрачена або набута, тим значніші відхилення у розвитку, тим раніше вони проявляються в онтогенезі. Тому аномалії за крупними хромосомами зустрічаються дуже рідко. Крім цього, нестача генетичного матеріалу відображається на організмі важче, ніж його надлишок і тому повні моносомії (особливо у живонароджених) зустрічаються значно рідше, ніж повні трисомії. Клінічна картина залежить не тільки від розміру хромосоми, яка бере участь в патологічному процесі, але й велике значення має і її якісний склад.
Синдром Дауна
Синдром Дауна - трисомія за 21 хромосомою - генетична аномалія, яку спричинює присутність додаткової хромосоми у 21 парі.
Синдром Дауна є хромосомною аномалією, якій притаманна додаткова хромосома 21 - цілковита (трисомія 21) або часткова (внаслідок транслокації).
Такі діти мають 47 хромосом у каріотипі замість звичних 46. Їм притаманна характерна зовнішність, підвищена можливість появи певного спектру захворювань (наприклад вроджених вад серцево-судинної системи, набутих вад сенсорної системи, тощо), певна розумова відсталість, через яку має місце повільніший розумовий розвиток на фоні здорових особин та гірша соціальна адаптація.
Через неможливість повної санації проблем даної аномалії медицина ставить на меті не постійне лікування, а забезпечення нормального соціального статусу для людей, що мають синдром Дауна.
За статистикою ВООЗ, у світі з синдромом Дауна народжується кожне 700-е немовля. Це співвідношення однакове в різних країнах, кліматичних зонах і соціальних прошарках. Генетичний збій відбувається незалежно від способу життя батьків, їхнього здоров'я, звичок і освіти.
Причини
Нажаль, дитина з синдромом Дауна може з'явитися в родині навіть у здорових людей. Генетика - річ складна і, в більшості випадків, непередбачувана, тому навряд чи варто важити на когось відповідальність за рішення природи.
Однак варто відзначити кілька факторів, які підвищують ризик народження в родині людини з синдромом Дауна:
• Пізні пологи. На відміну від молодих матусь, жінки старше 35 років більше схильні до ризику народження хворої дитини.
|
Вік матері |
Ступінь ризику |
|
від 20 до 24 років |
1 до 5000 |
|
від 25 до 30 років |
1 до 1000 |
|
від 35 до 39 років |
1 до 214 |
|
після 45 років |
1 до 19 |
• Вік батька. Цей фактор впливає в меншій мірі, ніж вік матері, але погіршення якості сперми з віком, орієнтовно після 42 років, теж підвищує ризики генетичних дефектів.
• Брак між кровними родичами. У людей зі схожою генетичною інформацією ймовірність прояву генетичних відхилень дуже велика.
• Спадковість. Ризик передачі захворювання у спадок, від близьких родичів, вкрай малий, але можливий при деяких різновидах синдрому.
• Шкідливі звички. Куріння, алкоголь, наркотики, якими зловживають майбутні батьки, можуть істотне вплинути на їх генетичний матеріал і здоров'я майбутнього малюка.
Причини народження дітей з синдромом Дауна часом криються у спадковості. За даними безлічі досліджень, синдром в більш ніж 90% випадків представлений класичною трисомією 21 хромосоми. Ця форма захворювання не передається у спадок. Однак відзначено, що в сім'ях, де народжувався малюк з синдромом Дауна, ймовірність виникнення хромосомної аномалії у наступну дитину вище, ніж у загальній популяції.
При транслокаційний формі синдрому дитина може отримати хвороба у спадок від здорових батьків. В даному випадку, один з батьків - носій генетичного матеріалу, в якому дві маленьких хромосоми зчіплюються в одну. Подібні обміни називаються збалансованою хромосомною перебудовою. У процесі перебудови кількість генетичного матеріалу не змінено, тому дані хромосомні зміни найчастіше не позначаються на здоров'ї самого носія перебудови. Однак при передачі подібного генетичного матеріалу наступному поколінню, перебудова може прийняти статус незбалансованою. Синдром Дауна розвивається в разі, якщо гени з 21 хромосоми будуть незбалансовані в бік збільшення їх кількості.
Мозаїчна форма захворювання не має тенденції до спадковості. Вона з'являється внаслідок випадкових змін в процесі мейозу на ранніх стадіях розвитку плоду. Результатом цих змін стає стан, в якому частина клітин тіла містить покладені дві копії 21 хромосоми, а в інших клітинах буде спостерігатися по три копії.
Трисомія 21. Абсолютна більшість випадків виникнення синдрому Дауна відбувається внаслідок потроєння 21 хромосоми (трисомія 21). Іншими словами, в кожній з клітин організму присутні три примірника даної хромосоми, коли має бути виключно дві копії. Порушення коректного складу генної інформації відбувається в батьківському організмі на етапі становлення статевих клітин. У 90% випадків це трапляється через збої в материнській яйцеклітині, інші випадки пояснюються особливостями сперматозоїдів. На стадії мейозу (поділ клітин) існує ймовірність того, що відбудеться помилка, через яку хромосоми не розійдуться до різних полюсів. Наслідком цього буде зміст надлишкової копії у клітині (замість набору з 23 хромосом клітина буде містити збільшений комплект з 24 хромосом). У разі, коли така статева клітина стає учасником процесу запліднення, у плода буде 47 хромосом замість 46. Через настільки ранню появу аномалії залучаються абсолютно всі клітини організму.
Мозаїчна форма синдрому. На більш пізній стадії також можуть трапитися порушення генетичного коду, проте ймовірність цього вкрай мала - від 1-2 до 5%. В такому випадку частина клітин зародка матиме нормальну кількість хромосом, а в якійсь одній (або кількох) з'явиться їх надмірна кількість. Ця клітина в подальшому дасть початок клону клітин із зайвою хромосомою. У результаті, частина клітин в організмі виявиться з надлишками генетичних елементів, а частина - з нормальним хромосомним набором. Мозаїчна форма - визначення, яким назвали дану аномалію. Тяжкість наслідків для організму в разі мозаїчної форми не настільки очевидна, як у випадку трисомії 21, адже зміни відбуваються лише в окремих ділянках організму. Виявити наявність даної форми захворювання набагато важче, особливо за допомогою пренатальних методів діагностики. Виразність клінічних симптомів залежить від співвідношення кількості нормальних клітин і клітин з аномальним хромосомним набором.
Робертсонівські транслокації. В даному випадку генотип хворого не буде відрізнятися від генотипу здорової людини. Транслокації називають вид мутацій, який має на увазі зміни в структурі хромосом. У разі роберсонівської транслокації 21 хромосома прикріплюється до іншої хромосоми, в більшості випадків до 14-ї в каріотипі одного з батьків. У загальній кількості випадків встановлення діагнозу «синдром Дауна» робертсонівські транслокації, як причина патології, займають всього 2-3%.
Існує така точка зору, що придбані мутації (опромінення іонізуючим випромінюванням і інші мутагенні фактори) також можуть викликати генетичні порушення, що сприяють виникненню синдрому Дауна. Глибокого дослідження щодо цього питання на на сьогоднішній день не проводилося, тому робити однозначні висновки з приводу цієї версії походження патології було б неправильно.
Клінічні ознаки
Особи з синдромом Дауна можуть мати деякі або всі з наступних ознак:
косі розрізи очей, з епікантусними складками (внутрішній кут ока),
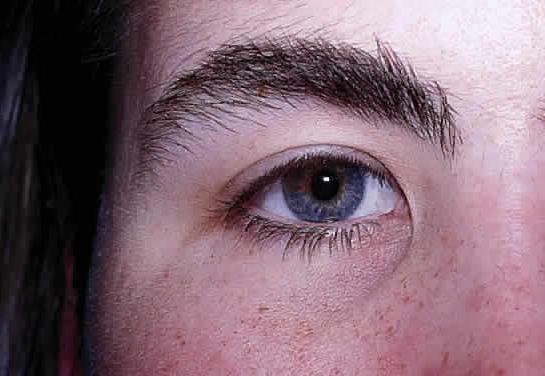
Рис. 14 – Епікантусні складки
гіпотонію м'язів,
пласке перенісся,
виступаючий язик як наслідок малого розміру ротової порожнини,
коротку шию,
білі плями на рогівці (так звані плями Брашфілда),
надмірну гнучкість суглобів, включно з атланто-аксіальним,
вроджені вади серця,
надмірний проміжок між першим і другим пальцем стопи,
поодиноку згинальну складку мізинця і підвищену кількість дематогліфів з ліктьової сторони долоні.
Більшість людей із синдром Дауна мають розумове відставання (IQ: 35-70), особи з мозаїчним синдромом Дауна мають дещо ліпші розумові здатності — на 30-40 пунктів вище.
пласке обличчя, на якому слабо виділяються ніс, рот, надбрівні дуги і тд;
брахицефалия (занадто укорочений череп), часто супроводжується плоским потилицею;
складка шкіри на шиї у новонароджених;
Деякі з цих характеристик можна побачити у ще не народженого малюка на УЗД. Однак з точністю визначити наявність синдрому Дауна по УЗД неможливо. Для цього лікар повинен призначити більш серйозні процедури дослідження.
Р
|
 ис.
15 –Зовнішній вигляд дітей з синдромом
Дауна
ис.
15 –Зовнішній вигляд дітей з синдромом
Дауна
• Маленький ніс.
• Коротка і широка шия.
• Збільшені очі, часто зустрічається косоокість.
• Укорочені кінцівки помітно не відповідають величині тіла.
• Невелика довжина кисті, викликана недорозвиненістю середніх пальців рук.
• Викривлена форма мізинця.
|
|
|
Рис. 16. – «Мавп`яча» борозна |
Порушення нормального функціонування систем організму також породжує деякі зовнішні особливості. Зокрема, у хворих нерідко спостерігається гіперрухливість суглобів і недостатній м'язовий тонус. Дані стану спостерігаються приблизно у 80% випадків. Через збільшений язик (макроглосія) і своєрідної будови піднебіння, рот хворого завжди відкритий. М'язи особи не здатні утримувати губи в закритому положенні. У 65% випадків виявляються аномалії зубів.
Характерні риси розумового розвитку:
• Помітне відставання у розвитку. Навіть при своєчасному комплексному лікуванні затримки в розвитку будуть все помітніше з віком. Розвиток розумових здібностей, в більшості випадків, залишається на рівні семирічної дитини. У рідкісних випадках інтелект досягає більш високого рівня.
• Невеликий словниковий запас. Крім того, що пацієнти з даною аномалією невиразно говорять, відзначається досить мізерний мовний набір.
• Відсутність здібностей до абстрактного мислення. Таким дітям легше розуміти і аналізувати тільки те, що вони бачать безпосередньо перед очима, уявити якусь ситуацію або пофантазувати для них вже проблематично.
• Низька концентрація уваги. Дитині важко сфокусуватися на будь-яку задачу, досить швидко вони починають відволікатися.
Синдром Дауна - це практично єдина хромосомна аномалія, коли діагноз можна поставити клінічно, тобто, орієнтуючись тільки на зовнішні ознаки. Однак в будь-якому випадку необхідно буде провести каріотипування, щоб визначити форму синдрому.
Окрім наявності самого синдрому Дауна у хворих практично завжди відзначається ряд інших проблем зі здоров'ям. Носії цієї хромосомної аномалії дуже часто стикаються з недугами кардіологічного характеру. Вроджені вади серця відзначаються приблизно в 40% випадків. Порушення біологічних процесів тягне за собою також ранній розвиток катаракти, з яким стикаються дві третини хворих старше восьми років. При синдромі Дауна значно збільшується ризик розвитку хвороби Альцгеймера і появи гострого мієлоїдного лейкозу. У пацієнтів з даною патологією часто є відхилення в роботі шлунково-кишкового тракту. Рекомендуються регулярні обстеження у гастроентеролога і кардіолога.
Простудні захворювання, ГРВІ та пневмонії - регулярні супутники хворого синдромом Дауна. Це пояснюється ослабленою імунною системою організму.
Поява зайвої хромосоми в організмі викликає також порушення обміну речовин, що призводить до несправного функціонування внутрішніх органів (захворювання щитовидної залози, порушення зору, слуху і т.д).
Треба зауважити, що результати деяких досліджень вказують на те, що люди, які є носіями цієї хромосомної аномалії, мають відносно невисоку частоту виникнення злоякісних пухлин в організмі. Однак накопичена на сьогоднішній день інформація щодо даного медичного питання не дає змоги з упевненістю говорити про причини такого взаємозв'язку.
Лікування
Генетична природа захворювання зводить шанси на успішну боротьбу з ним до мінімуму. Однак імовірність того, що все-таки будуть розроблені методики, що дозволяють проводити успішне лікування синдрому Дауна. Дослідження вчених, проведені в останні роки, показали, що можна зробити своєрідне "виключення" зайвої копії 21 хромосоми. Щоб здійснити дану дію, передбачається використовувати особливу поведінку X-хромосоми, яка відповідає за приналежність людини до жіночої статі. Чоловічим набором хромосом, як відомо, є XY, а жіночим - набір XX. Вченими було встановлено, що в організмі жінки у одній з хромосом спостерігається інактивований стан, а її дані не використовуються в процесі синтезу білкових структур. По суті, вона не є учасником системи біохімічних реакцій в організмі. Відбувається це внаслідок вироблення особливих речовин під керуванням специфічної ділянки ДНК. Ці речовини покривають поверхню хромосоми і, тим самим, блокують доступ до неї.
Медики зробили спробу скористатися цим механізмом, з метою зробити неактивною третю копію 21 хромосоми. Дані досліджень показують, що саме її активність в процесі виробництва білкових з'єднань ініціює появу різноманітних відхилень від норм розвитку. Для реалізації задумки необхідно було зробити зайвий екземпляр 21 хромосоми схожим на X-хромосому. Для цього вчені застосували кілька модифікованих ділянок генетичного матеріалу, що викликає інактивацію. Ця ділянка була поставлена замість одного з «сміттєвих» фрагментів (частина ДНК, яка відповідає за синтез білків) в зайву хромосому. Через деякий час, оброблена подібним чином копія 21 хромосоми припинила проявляти активність. Дослідники висунули припущення, що це дозволить компенсувати наявність зайвого генетичного матеріалу.
Необхідно зауважити, що досліди проводилися тільки на культурі стовбурових клітин, а значить, на сьогоднішній день неможна говорити про будь-яку ефективність методу. Крім того, допомога від цього підходу можуть отримати тільки ті люди з синдромом Дауна, у яких він обумовлений трисомією. Транслокаційну форму хвороби подібним чином вилікувати неможливо: довелося б «вимикати» цілу хромосому, а це однозначно принесе більше шкоди, ніж користі. Однак, з огляду на стрімкий прогрес в сфері генетики та пов'язаних з нею досліджень, можна сказати, що метод має певні перспективи.
Соціалізація і інтеграція в суспільство
Дітей з синдромом Дауна іноді називають «сонячними». Пояснюється це тим, що вони дуже доброзичливі і позитивні, не дивлячись на можливі перепади настрою і періодичні спалахи агресії. Завдяки стрімкому розвитку медицини і педагогіки у малюків з синдромом Дауна тепер набагато більше перспектив у отриманні освіти, ніж це було на початку XX сторіччя. В ті часи тривалість життя людей з даною патологією становила в середньому 20-25 років. Вплив цього фактора не дозволяв їм приділяти достатню кількість часу освоєння тих чи інших дисциплін. Останні статистичні дослідження на питання «скільки живуть люди з синдромом Дауна?» Відповідають, що планка тривалості життя піднялася до 50 років.
Синдром Едвардса
Синдром Едвардса або трисомія 18 являє собою важке вроджене захворювання, викликане хромосомними порушеннями. Воно є однією з найбільш поширених патологій в даній категорії (поступається за частотою лише синдрому Дауна). Захворювання характеризується численними порушеннями у розвитку різних органів і систем. Прогноз для дитини зазвичай несприятливий, але багато чого залежить від догляду, який здатні забезпечити батьки.
Поширеність синдрому Едвардса по земній кулі варіює від 0,015 до 0,02%. Чіткої залежності від місцевості або раси не спостерігається. Статистично дівчатка хворіють у 3 – 4 раза частіше хлопчиків. Наукового пояснення цієї пропорції поки не виявлено. Тим не менш, відзначено ряд факторів, які можуть підвищити ризик виникнення цієї патології.
Як і інші хромосомні мутації, синдром Едвардса є, в принципі, невиліковним захворюванням. Найсучасніші методи лікування та догляду можуть лише підтримувати життя дитини і сприяти певному прогресу в його розвитку. Єдиних рекомендацій по догляду за такими дітьми немає через величезне розмаїття можливих порушень і ускладнень.
Причини
Синдром Едвардса є генетичним захворюванням, яке характеризується наявністю додаткової хромосоми в геномі людини.
Синдром Едвардса відноситься до так званих хромосомних захворювань, коли проблема полягає не в дефекті гена, а в дефекті цілої молекули ДНК. Якщо бути більш точним, то класична форма цього захворювання - наявність зайвої 18-ї хромосоми. Каріотип в таких випадках позначається як 47, ХХ, 18+ (для дівчинки) і 47, ХY, 18+ (для хлопчика). Остання цифра позначає номер додаткової хромосоми. Надлишок генетичної інформації в клітинах призводить до появи відповідних проявів хвороби, які і об’єднані під назвою «синдром Едвардса». Наявність додаткової (третьої) хромосоми під номером 18 дало іншу (більш наукову) назву хвороби – трисомія 18.
Залежно від форми хромосомного дефекту розрізняють три види даного захворювання:
Повна трисомія 18. Повна або класична форма синдрому Едвардса припускає, що всі клітини в організмі мають додаткову хромосому. Даний варіант захворювання зустрічається більш ніж в 90% випадків і є найбільш важким.
Часткова трисомія 18. Часткова трисомія 18 є досить рідкісним феноменом (не більше 3% від усіх випадків синдрому Едвардса). При ній у клітинах організму міститься не ціла додаткова хромосома, а лише її фрагмент. Такий дефект може бути результатом неправильного поділу генетичного матеріалу, але зустрічається він дуже рідко. Іноді частина вісімнадцятої хромосоми приєднується до іншої молекули ДНК (впроваджується в її структуру, подовжуючи молекулу, або просто «чіпляється» за допомогою містка). Подальший поділ клітин призводить до того, що в організмі є 2 нормальні хромосоми номер 18 і ще частина генів з цих хромосом (зберігся фрагмент молекули ДНК). У цьому випадку кількість вроджених дефектів буде набагато нижче. Спостерігається надлишок не всієї генетичної інформації, закодованої у 18-й хромосомі, а лише її частини. Для пацієнтів з частковою трисомією 18 прогноз краще, ніж для дітей з повною формою, але все одно залишається несприятливим.
Мозаїчна форма. Мозаїчна форма синдрому Едвардса зустрічається в 5 – 7% випадків даного захворювання. Механізм її появи відрізняється від інших видів. Справа в тому, що тут дефект утворився вже після злиття сперматозоїда і яйцеклітини. Обидві гамети (статеві клітини) спочатку мали нормальний каріотип і несли по одній хромосомі кожного виду. Після злиття сформувалася клітина з нормальною формулою 46, ХХ або 46, XY. У процесі поділу цієї клітини стався збій. При подвоєнні генетичного матеріалу один з фрагментів отримав додаткову 18-ю хромосому. Таким чином, на певному етапі сформувався зародок, частина клітин якого мають нормальний каріотип (наприклад, 46, ХХ), а частина – каріотип синдрому Едвардса (47, ХХ, 18+). Частка патологічних клітин ніколи не перевищує 50%. Їх число залежить від того, на якому етапі поділу початкової клітини стався збій. Чим пізніше це відбувається, тим менше буде частка дефектних клітин. Форма отримала назву через те, що всі клітини організму являють собою своєрідну мозаїку. Частина з них здорова, а частина – з важкою генетичною патологією. Закономірності в розподілі клітин в організмі при цьому не спостерігається, тобто всі дефектні клітини не можуть локалізуватися тільки в одному місці, щоб їх можна було видалити. Загальний стан пацієнта при цьому легше, ніж при класичній формі трисомії 18.
Наявність додаткової хромосоми в геномі людини представляє безліч проблем. Справа в тому, що клітини людини запрограмовані зчитувати генетичну інформацію і дублювати лише задану природою кількість молекул ДНК. Порушення навіть у структурі одного гена можуть призвести до серйозних захворювань. При наявності ж цілої молекули ДНК розвиваються множинні порушення ще на етапі внутрішньоутробного розвитку до народження дитини.
Згідно з останніми дослідженнями хромосома номер 18 містить 557 генів, які кодують не менше 289 різних білків. У відсотковому відношенні це приблизно 2,5% всього генетичного матеріалу. Порушення, які викликає настільки великий дисбаланс, дуже серйозні. Неправильна кількість білків зумовлює безліч аномалій у розвитку різних органів і тканин. У разі синдрому Едвардса частіше за інших страждають кістки черепа, деякі відділи нервової системи, серцево-судинна і сечостатева система. Скоріше за все, це пов’язано з тим, що гени, розташовані на цій хромосомі, мають відношення до розвитку саме цих органів і систем.
Таким чином, основною і єдиною причиною синдрому Едвардса є наявність додаткової молекули ДНК. Найбільш часто (при класичній формі хвороби) вона успадковується від одного з батьків. У нормі кожна гамета (сперматозоїд і яйцеклітина) містять по 22 непарні соматичні хромосоми, плюс одна статева. Жінка завжди передає дитині стандартний набір 22 + Х, а чоловік може передати 22 + Х або 22 + Y. Це зумовлює стать дитини. Статеві клітини батьків утворюються в результаті поділу звичайних клітин на два набори. У нормі материнська клітина ділиться на дві рівні частини, але іноді не всі хромосоми діляться навпіл. Якщо 18-а пара не розійшлася по полюсам клітини, то одна з яйцеклітин (або один з сперматозоїдів) буде заздалегідь дефектним. У ньому буде не 23, а 24 хромосоми. У разі якщо саме ця клітина буде брати участь у заплідненні, дитина отримає додаткову 18-у хромосому.
На неправильний поділ клітин можуть вплинути наступні фактори:
-
Вік батьків. Доведено, що ймовірність хромосомних аномалій збільшується прямо пропорційно з віком матері. При синдромі Едвардса цей зв’язок менш виражений, ніж при інших схожих патологіях (наприклад, синдром Дауна). Але для жінок старше 40 років ризик народити дитину з даною патологією в середньому в 6 – 7 разів вище. Подібна залежність від віку батька спостерігається в значно меншому ступені.
-
Куріння і алкоголь. Такі шкідливі звички як куріння і зловживання алкоголем можуть діяти на статеву систему людини, впливаючи на розподіл статевих клітин. Таким чином, регулярне вживання цих речовин (а також інших наркотичних препаратів) підвищує ризик неправильного розподілу генетичного матеріалу.
-
Прийом лікарських засобів. Деякі лікарські препарати при неправильному прийомі в першому триместрі можуть вплинути на розподіл зародкових клітин і спровокувати мозаїчну форму синдрому Едвардса.
-
Захворювання статевої сфери. Перенесення інфекції з ураженням репродуктивних органів можуть відбитися на правильному розподілі клітин. Вони підвищують ризик хромосомних і генетичних захворювань в цілому, хоча спеціально для синдрому Едвардса подібні дослідження не проводилися.
-
Радіаційне випромінювання. Опромінення статевих органів рентгенівським випромінюванням або іншими іонізуючими випромінюваннями може спричинити генетичні мутації. Особливо небезпечно таке зовнішній вплив в підлітковому віці, коли поділ клітин відбувається найбільш активно. Частинки, що формують випромінювання, легко проникають крізь тканини і піддають молекулу ДНК своєрідному «бомбардуванню». Якщо це відбувається в момент поділу клітини, ризик хромосомної мутації особливо високий.
В цілому ж можна сказати, що причини розвитку синдрому Едвардса остаточно відомі і добре вивчені. Вищеперелічені чинники лише підвищують ризик розвитку даної мутації. Не виключається і вроджена схильність деяких людей до неправильного розподілу генетичного матеріалу в статевих клітинах. Наприклад, вважається, що у подружньої пари, яка вже народила дитину з синдромом Едвардса, ймовірність появи другої дитини з аналогічною патологією складає аж 2 – 3% (приблизно в 200 разів вище, ніж середня поширеність цієї хвороби).
Клінічні ознаки
|
Р |
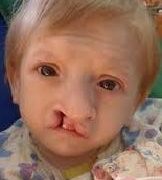 ис.
17 – Зовнішній вигляд дитини хвору на
синдром Едвардса
ис.
17 – Зовнішній вигляд дитини хвору на
синдром ЕдвардсаНовонароджені діти з синдромом Едвардса зазнають труднощів з ковтанням і як наслідок цього виникають проблеми з годуванням. З перших тижнів відзначається затримка фізичного розвитку. Через грубі вади розвитку центральної нервової системи діти страждають розумовою відсталістю.
З боку серцево-судинної системи спостерігаються вади розвитку головних кровоносних судин і серцевого м'яза, що стає основною причиною смерті немовлят в перші місяці життя. У нирках також спостерігаються стійкі порушення, найчастіше це гидронефроз, тобто прогресуюче розширення порожнин нирок з наступною атрофією ниркової тканини. Статеві органи дітей з синдромом Едвардса недорозвинені.
З боку шлунково-кишкового тракту у пацієнтів відзначається зрощення вивідних проток жовчних ходів і жовчного міхура, звуження просвіту стравоходу і килеподібне випинання кишечника.
Діагностика
Діагностика цього синдрому полягає у проведенні хромосомних тестів. Під час перебігу вагітності, навіть не дивлячись на те, що весь період виношування проводиться УЗД плода - діагностика синдрому Едвардса найбільш ускладнена. Дані ультразвукового дослідження вказують лише на непрямі ознаки, які надалі можуть привести до синдрому Едвардса (недорозвинення або відсутність у пупковому каналі пупкової артерії, невелика величина плаценти). У перші три місяці перебігу вагітності за допомогою ультразвукового дослідження неможливо виявити грубих аномалій плідного розвитку, які свідчать про наявність синдрому Едвардса. Тому внаслідок утрудненої діагностики цієї вади, питання про переривання вагітності у визначений для цього строк практично ніколи не виникає і жінки виношують і народжують дітей з синдромом Едвардса у визначений термін
Лікування
На сьогоднішній день способів виправлення хромосомних порушень не існує, тому внаслідок наявності численних вад фізичного та психічного розвитку, більшість дітей (понад 90%) помирає в перший рік життя, причому близько 30% протягом першого місяця. Одиниці які вижили, протягом усього свого досить недовгого життя будуть страждати численними соматичними захворюваннями та глибокою розумовою відсталістю.
Синдром Патау
Трисомія 13 або синдром Патау - мутація, патологія, генне захворювання, при якому в клітинах з'являється додаткова тринадцята хромосома. Стоїть в одному ряду з такими захворюваннями, як синдром Дауна або Едвардса. Однак зустрічається набагато рідше їх і проявляється цілим набором зовнішніх каліцтв і внутрішніх патологій. У медицині можна зустріти інші назви хвороби: трисомія D, синдром Патау-Смітта. Про причини цієї мутації генетики можуть тільки здогадуватися.
Причини
Основні причини синдрому Патау, виявлені на сьогоднішній день:
• помилки у формуванні сперматозоїдів і яйцеклітин, що беруть участь у зачатті дитини, тобто абсолютно випадковий фактор;
• вік матері після 45 років;
• несприятлива екологія, особливо при радіаційному зараженні;
• шлюби між родичами;
• спадковість - рідкісна причина, так як генетичний матеріал несе в собі здорова людина, що не має ознак трисомії 13.
Через це в хромосомному наборі дитини можуть виникнути патології. Враховуючи цей список причин, молоді матусі повинні уникати цих чинників, наскільки це можливо. А при попаданні в групу ризику пройти всі необхідні дослідження для виявлення захворювання. Симптоматика зумовлена тим, що в основі синдрому Патау лежить наявність додаткової хромосоми 13, якої немає у звичайних людей.
Симптоми і ознаки
Існують різні ознаки синдрому Патау, які не сплутаєш ні з якою іншою хворобою. Так як це геномна мутація, її симптоми яскраво виражаються навіть у зовнішності. Вроджені вади нерідко призводять до летального результату дітей ще в дитинстві. Так як в наявності зовнішні каліцтва, батьки часто відмовляються від таких діток.
Явні УЗД-ознаки синдрому Патау:
• зовнішні каліцтва;
• уповільнений розвиток плода;
• тахікардія (в 70% випадків);
• багатоводдя (в 50% випадків) - ця ознака дозволяє виявити синдром Патау на УЗД з урахуванням інших діагностик;
• мегацістіс (збільшений в розмірах сечовий міхур);
• голопрозенцефалія - нерозділення мозку на дві півкулі;
• омфалоцеле - ембріональна пуповинна грижа.
Зовнішні симптоми (рис.18):
Р
|
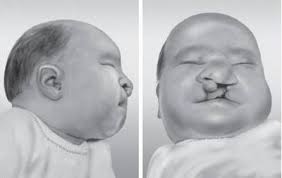 ис.
18 – Зовнішні симптоми дитини з синдромом
Патау
ис.
18 – Зовнішні симптоми дитини з синдромом
Патау
• помірна мікроцефалія - зменшені розміри черепа і головного мозку;
• низький лоб, найчастіше скошений;
• вузькі очні щілини на невеликій відстані один від одного;
• мікроофтальмія - недорозвинення очей;
• колобома - відсутність очної оболонки;
• запале перенісся;
• помутніння рогівки;
• деформовані вушні раковини;
• ущелини піднебіння;
• полідактилія - більша кількість пальців на руках і ногах, ніж у інших людей (рис.19);
• вигин кистей;
• коротка шия.
|
Р |
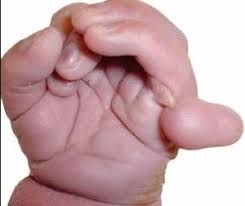 ис.
19 - Полідактилія
ис.
19 - Полідактилія• Порушення функціонування відділів ЦНС;
• вади розвитку серця (у 80%): дефекти перегородок (міжшлуночкової і міжпередсердної), транспозиції судин;
• додаткова селезінка;
• фіброкістозні зміни в підшлунковій залозі;
• ембріональна пупкова грижа;
• збільшені нирки;
• дольковість нирок;
• наявність в кірковому шарі нирок кісти;
• патології статевих органів: їх гіпоплазія, у хлопчиків крипторхізм - неопущення яєчок, у дівчаток - дворога матка;
• затримка розумового розвитку;
• відсутність задньої стінки сечівника.
Основні, типові ознаки захворювання виявляються при УЗД вже на 12 тижні вагітності, а потім підтверджуються всілякими діагностичними дослідженнями. Якщо вони не проводилися пренатально, клінічна картина синдрому Патау після народження малюка досить яскраво ілюструє саме цей діагноз. Але для виключення помилки проводяться аналізи на каріотип. У генетиці розрізняють кілька типів патології.
Класифікація
Розрізняють два цитогенетичних виду синдрому Патау:
• проста трисомія;
• робертсонівські транслокация.
Набагато рідше діагностуються інші цитогенетичні різновиди захворювання:
• мозаїцизм - наявність в тканинах людини генетично різних клітин;
• ізохромосома;
• неробертсонівська транслокація.
Клінічна картина простих трисомних і транслокаційний форм не відрізняється. Всі клітини дитини мають характерний каріотип синдрому Патау - 47, XX 13+ або 47, XY 13+.
Поряд з іншими схожими захворюваннями, синдром Патау - це генетична мутація, природу якої ученим ще тільки належить з'ясувати. Однак від тих же синдромів Дауна, Шерешевського-Тернера, Едвардса, котячого крику, Клайнфельтера дану патологію відрізняють такі особливості:
• згідно з останніми даними, частота народження синдрому Патау - один випадок на 10 000;
• між цим показником віком матері простежується нечітка, але все-таки залежність, вона менш сувора, ніж при народженні дітей з синдромом Дауна;
• малюки народжуються з пренатальної гіпоплазією (недорозвиненістю тканин і органів), яка не пояснюється недоношенностью (середній термін таких вагітностей становить зазвичай близько 38,5 тижнів);
• зовнішні і внутрішні каліцтва не дозволяють таким дітям вести нормальний спосіб життя (рис.20).
Незважаючи на те, що сьогодні ведеться активна пропаганда проти абортів, а синдром Патау - хвороба дуже рідкісна, якщо вона була виявлена на ранніх термінах вагітності, лікарі однозначно порадять її екстрене переривання. Це позбавить і батьків, і ще не народженого малюка від безлічі мук.
Діагностика
Важливу роль відіграє діагностика синдрому Патау, так як вона дозволяє відрізнити захворювання від подібних генних мутацій (синдрому Меккеля і Мора, трігоноцефалії Опітц), які за деякими ознаками збігаються з трисомєю 13. Вирішальним фактором є цитогенетичне дослідження хромосом. Воно необхідне для прогнозу життя та здоров'я дітей, дозволяє своєчасно перервати вагітність при підтвердженні діагнозу.
I етап
1. УЗД-дослідження.
2. Визначення біохімічних і фізичних маркерів бета-ХГЧ (гормону хоріона), РАРР-А (білка плаценти) і ін.
3. Розрахунок шансів на народження малюка з синдромом Патау.
II етап
На даному етапі діагностика проводиться для тих вагітних, хто потрапив в групу ризику за підсумками досліджень I етапу.
1. 8-12 тижнів: береться біопсія хоріона;
2. 14-18 тижнів: проводиться амніоцентез - прокол амніотичної оболонки, щоб дослідити навколоплідні води;
3. 20 тижнів: кордоцентез - дослідження пуповинної крові.
У матеріалах, отриманих в ході цих заходів, генетики шукають патологію методом КФ-ПЛР або за допомогою каріотипування, диференційно фарбуючи хромосоми. Якщо діагностика при вагітності не проводилася, хромосомну аномалію виявляють на підставі клінічної картини. Для точної постановки діагнозу обов'язковий генетичний аналіз для визначення каріотипу новонародженого.
Лікування
Якщо батьки вирішили залишити дитину з синдромом Патау, їм належить ретельна діагностика і лікування аж до кінця життя малюка. Але так як неможливо виправити хромосомні порушення, просто ведеться комплексна робота різних фахівців: педіатра, генетика, невролога, кардіолога, офтальмолога, ортопеда-травматолога, отоларинголога, гастроентеролога, уролога та ін. Вона полягає в:
• численних обстеженнях, спрямованих на виявлення вад розвитку: до них відносяться нейросонографія (ультразвукове секторальне сканування головного мозку новонародженого через тім'ячко), УЗД органів черевної порожнини нирок, ехокардіографії (ультразвукове дослідження серцевих патологій) і ін .;
• постійному контролі за здоров'ям дитини;
• оперативних втручаннях для корекції деяких вроджених вад;
• ретельного догляду за дитиною;
• загальнозміцнюючу терапії для нормального функціонування пошкоджених органів і систем: це полівітамінні комплекси, зміцнення імунітету, біологічно активні добавки;
• запобігання інфекційних ускладнень у малюка;
• психологічна підтримки сім'ї.
Так як генетика - основна причина народження дітей з синдромом Патау, навіть найостанніші розробки медицини не вступають з нею в боротьбу. Тому лікування цього захворювання не вносить кардинальних змін в стан хворих.
|
Р |
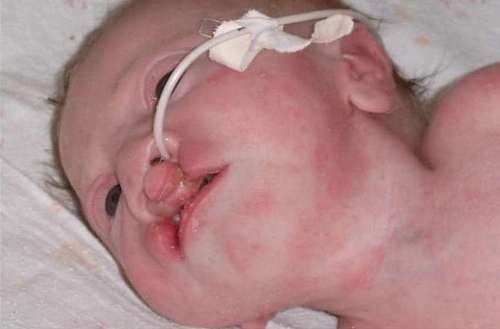 ис.
20 – Зовнішній вигляд дитини з синдромом
Патау
ис.
20 – Зовнішній вигляд дитини з синдромом
ПатауЧерез важких вроджених вад розвитку прогнози для дітей з синдромом Патау невтішні. Зокрема:
• викидні;
• висока загроза мертвонародження;
• 95% новонароджених помирають до року, в перші тижні або місяці після народження;
• решта 5% живуть ще кілька років;
У розвинених країнах останнім часом намітилася позитивна тенденція: тривалість їхнього життя збільшується; 15% - до 5 років, 2-3% - до 10 років.
Діти з синдромом Патау, що залишилися в живих, до самої смерті страждають глибокою формою ідіотії, залишаються повними інвалідами. Якщо причина народження такої дитини - випадковий фактор, такі вагітності можуть виключити повторний розвиток подій. Якщо вся справа в робертсонівській транслокації, здоровий малюк у її носія навряд чи народиться.
Профілактика
Через неточне визначення причин синдрому Патау специфічних методів його профілактики не існує. Правда, можна дати кілька корисних порад, які знизять ризик розвитку цього захворювання:
• змінити район проживання, якщо він відрізняється несприятливою екологією;
• уникати радіаційного зараження і контакту з шкідливими хімічними речовинами;
• не народжувати дітей від близьких кровних родичів;
• при наявності спадкового подібного захворювання обов'язково пройти медико-генетичне консультування, бажано - ще до зачаття, на етапі планування вагітності;
• не народжувати після 45 років.
Синдром Патау - важке генетичне захворювання, хромосомна мутація, яка дуже ускладнює життя дитини. Якщо батьки не наважилися перервати вагітність або з якоїсь причини діагноз не був поставлений своєчасно і малюк народився, їм батькам доведеться чимало сил і часу на те, щоб хоч трохи продовжити життя свого малюка-інваліда. Так, він ніколи (навіть після безлічі хірургічних і пластичних операцій) не буде таким, як інші дітки, але для своєї мами він завжди буде найкращим і бажаним.
Синдром Шерешевського –Тернера
Синдром Шерешевського-Тернера - це захворювання генетичного спадкового характеру, в результаті якого відбуваються порушення в структурі Х-хромосоми, що супроводжуються аномаліями розвитку внутрішніх органів і низькорослістю. Це як спадкове захворювання описано у 1925 році ендокринологом Шерешевским, на думку якого, воно обумовлено не повним розвитком гіпофіза у передній частці і статевих залоз з одночасними вродженими вадами соматичного характеру. А ось вже Тернером у 1938 році були виділено додаткові три симптому до загальних ознак захворювання. До них відносяться деформації ліктьових суглобів, наявні на шкірі шкірні складки у вигляді крил і статевий інфантилізм.
Причини
Хромосомна патологія плода є основою причини виникнення даного захворювання, яке призводить до важкої вагітності та передчасних пологів, у результаті чого народжується дитина з синдромом Шерешевського-Тернера. Ця аномалія, як виявлено, не залежить від віку або якихось патологічних захворювань батьків. Тому набір патологічних хромосом становить основу синдрому. Цей дефект виникає після нерозходження хромосом матері або батька.
В організмі здорової людини знаходиться сорок шість хромосом, а хворі з синдромом Шерешевського-Тернера, не мають однієї хромосоми, її просто не існує. Замість подвійної ХХ хромосоми, які властиві жінкам, міститься тільки одна Х-хромосома сорок п'ята (ХО). Якщо ця хромосома повністю відсутня або піддається змінам, то порушується утворення ферментів і білків в організмі, що призводить до загального дисбалансу. Така патологія є однією з цитогенетичних форм синдрому Шерешевського-Тернера. Але найбільш частіше зустрічається другий варіант - це мозаїцизм, який характеризується структурними перебудовами ізохромосоми, що локалізується на довгому плечі Х-хромосоми. Тому, якщо головна причина розвитку синдрому Шерешевського-Тернера - це порушення у каріотипі, то вже ці зміни можуть викликати різні впливу іонізуючих випромінювань на клітини під час ділення, шкідливі токсичні речовини і схильність організму на генетичному рівні до формування хромосом патологічної етіології.
Клінічні ознаки
Синдром Шерешевського-Тернера має основні клінічні і патофізіологічні особливості, що включають в себе, в першу чергу, порушення росту, передчасну оваріальную недостатність, вроджені вади з боку серцево-судинної та сечовидільної систем, скелетні дефекти, лімфатичні набряки рук і ніг, патології органів зору і слуху, а також метаболічні та фізіологічні зміни.
Практично у 95% випадків при синдромі Шерешевського-Тернера виявляється низькорослість. Кінцевий результат середньої величини зростання становить 140-147 см. Затримка в рості при даному синдромі обумовлена сукупністю дисплазії скелета з порушеннями хромосомах і внутрішньоутробному уповільненому зростанні.
Порушення, які відбуваються в гормінативному епітелії, призводять до первинної недостатності гонад та гонадобластомі. У 25% дівчаток з синдромом Шерешевського-Тернера зустрічається спонтанний пубертат, має мозаїчний варіант каріотипу. В основному він буває повним, тому і не дає можливості нормально і тривало функціонувати яєчникам.
Також пубертатний період характеризується відсутністю вторинних статевих ознак. Абсолютно не розвинені молочні залози, виявляється аменорея та мізерне оволосіння лобка, зовнішні статеві органи недорозвинені. Дуже рідко виявляються фолікули, що стає причиною неможливості дітонародження. Недолік естрогенів розвиває у жінок остеопороз, який викликає часті переломи шийки стегна, хребта, зап'ястя.
Дуже часто хворі синдромом Шерешевського-Тернера скаржаться на підвищення АТ. До того ж на ногах воно нижче норми або взагалі не визначається.
Основною причиною смерті хворих з синдромом Шерешевського-Тернера є розрив дилятованої аорти. Також у багатьох хворих виявляють коарктації аорти (провокує тиск) і бикуспідальний аортальний клапан.
Поширеним явищем при даному захворюванні є патології з боку сечовидільної системи. На УЗД виявляють вади розвитку у вигляді подвійної нирки і мальротації. Також зустрічається двостороння гіпоплазія нирок, змінюється кількість артерій і вен на них, подвоюються сечоводи і балії. Як правило, такі зміни не порушують функції сечовидільної системи, але призводять до артеріальної гіпертензії і стають причиною розвитку багатьох інфекцій.
При лімфостазі у хворих з синдромом Шерешевського-Тернера відзначаються набряки кистей і стоп, які з віком зникають, крильчасті складки на шиї (рис.21), дисплазія нігтів, аномалії у розвитку вушних раковин.
|
Р |
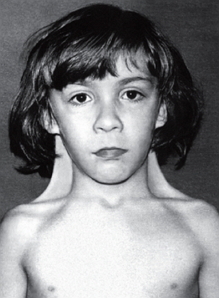 ис.
21 - Крильчасті складки на шиї
ис.
21 - Крильчасті складки на шиїПацієнти з синдромом Шерешевського-Тернера у своїй поведінці нагадують маленьких дітей, хоча міміка та вираз обличчя відображають дорослу людину.
Діагностика
Новонародженим дітям поставити діагноз синдром Шерешевського-Тернера досить складно, але вже після першого року життя, з появою фенотипових ознак, це стає цілком можливим. Для постановки діагнозу необхідно провести молекулярно-цитогенетичний аналіз, для виключення мозаїцизма. Діагностування синдрому Шерешевського-Тернера здійснюється в процесі дослідження каріотипу, виявлення соматичних аномалій та проведення лапаротомії. Носії даної патології знаходяться під регулярним наглядом онколога, так як нерозвинені гонади можуть перерости у дисгерміноми або гоноцитоми. В аналізі крові хворих на синдром Шерешевського-Тернера виявляють знижений кількість естрогенів при підвищених гормони гіпофіза (фоллитропіну). На УЗД - недорозвинення матки і відсутність яєчників. При рентгенологічному обстеженні виявляють остеопороз кісток і різного виду аномалії скелета. Дуже часто до основного захворювання приєднуються і інші хвороби внутрішніх органів.
Лікування
В першу чергу лікування синдрому Шерешевського-Тернера починають із застосування ростостимулюючої терапії. Це необхідно для того, щоб нормалізувати зростання в більш ранньому віці, індукувати пубертат теж в нормальному періоді і остаточно досягти істотних результатів у зростанні.
На сьогодні існує ефективний і безпечний препарат, що застосовується для лікування хворих з синдромом Шерешевського-Тернера - це рекомбінантний гормон росту (РГР). Генетики довели, що використання високих доз РГР дозволило збільшити зростання хворих до 157-163 см. В перший рік лікування відзначається максимальна швидкість в рості від 8 до 15 см, а потім відбувається її зниження до 5-6 см в рік.
Рано розпочате лікування синдрому Шерешевського-Тернера дає позитивний результат у соціально значущому кінцевому зростанні цих хворих.
Крім збільшеного зростання, при використанні рекомбінантного гормону, відзначається позитивна динаміка гормонального, психічного та метаболічного фону.
Одночасно з цим препаратом пацієнтам призначають соматотропін, який збільшує м'язову масу, покращує нирковий кровообіг, підвищує серцевий викид, збільшує всмоктування кальцію в кишечнику і збагачує мінералами кістки. В результаті цього в крові знижується рівень ліпопротеїнів, а рівні лужної фосфатази, жирних кислот, сечовини і фосфору збільшуються до норм. Пацієнти з синдромом Шерешевського-Тернера відчувають підвищення життєвого тонусу і їх життя значно поліпшується.
Індукцію пубертату проводять при використанні препаратів з естрогенами, які імітують нормальний статевий розвиток. Якщо раніше замісна терапія естрогенами розпочиналася з п'ятнадцяти років, щоб оптимізувати ростовий потенціал, то на даний момент за підсумковими даними Міжнародного консенсусу з лікування синдрому Шерешевського-Тернера, прийнято починати терапію естрогенами з дванадцяти років одночасно з РГР. Це пов'язано з перевіреною позитивною терапією цих гормонів. А ось при затримці пубертату, яка сприяє ранньому виявленню оваріальної недостатності, може посилитись негативний психологічний стан таких пацієнток.
У багатьох жінок з синдромом Шерешевського-Тернера після такого лікування з'являються шанси народити дитину. Адже після застосування гормонів росту збільшується до нормальних розмірів і матка, що дозволяє виносити дитину. Для цього використовується ЕКО з донорською яйцеклітиною.
В даний час розроблені різні корекційні методи лікування синдрому Шерешевського-Тернера. Наприклад, щоб врегулювати енергетичну систему організму застосовують голковколювання. Воно сприяє нормалізації обмінних процесів, покращує функції вегетативно-ендокринних органів, відновлює баланс всього організму. Голковколювання - це один з унікальних методів терапії, який покращує мікроциркуляцію біорідинами в системах і органах, допомагає краще функціонувати мозку і серцю, надає знеболюючий ефект в організмі.
При синдромі Шерешевського-Тернера є візуальний недолік на шиї у вигляді додаткових шкірних складок, які можна видалити хірургічним шляхом, зробивши пластичну операцію.
Для зняття задишки багатьом хворим призначають фізіотерапевтичні курси. У конкретному випадку - це інгаляції зволоженим киснем. Також дуже добре допомагає і лікувальна фізкультура, яка включає в себе певні вправи, спрямовані на конкретні групи м'язів (дихальні, руки або ноги). Ці вправи можуть бути як активними (виконуватися самим хворим), так і пасивними (з допомогою медичного працівника або, допомагаючи собі здоровою частиною тіла).
Завдяки такому комплексному лікуванню життя з синдромом Шерешевського-Тернера стає набагато краще.
Синдром Клайнфельтера
Описаний Н. F. Klinefelter et al. Ця хромосомна патологія зустрічається досить часто: вона виявляється в середньому 1:850 новонароджених чоловічої статі і в 1-2,8 % хворих на олігофренію, частіше при неглибокому інтелектуальному зниженні. Серед чоловіків, які страждають на безпліддя, більше 10 % мають додаткову Х-хромосому. Середній вік батьків при народженні дитини з хворобою Клайнфельтера підвищений: він дорівнює 35,5 років у батька.
Зовнішній вигляд немовлят із синдромом звичайний. Зміни, як правило, починають клінічно виявлятися в препубертатному і пубертатному віці.
Дорослі чоловіки мають високий зріст, євнухоподібну статуру (довгі ноги, високу талію, відносно широкий таз, відкладання жиру за жіночим типом), схильність до ожиріння, гінекомастію (рис.22). Як специфічний для захворювання симптом, який не зустрічається при інших формах гіпогонадизму, відзначають відносно короткі руки, їх розмах не більше ніж на 2-3 см перевищує зріст, у той час як при інших варіантах недостатності гормональної активності статевих залоз це розходження складає не менше 4 см. Даний симптом виявляється ще в допубертатному періоді. Пахвове оволосіння виражено недостатньо, на лобку оволосіння за жіночим типом, рослинність на обличчі незначна або відсутня.
|
Р |
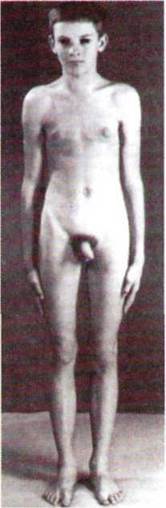 ис.
22 – Зовнішній вигляд хлопчиків з
синдромом
ис.
22 – Зовнішній вигляд хлопчиків з
синдромомСеред дерматогліфічних ознак нерідко зустрічаються поперечна складка, дистальне розміщення трирадіуса, збільшення частоти дуг на пальцях.
Розумова відсталість відмічається у 25-50 % хворих. Інтелектуальна недостатність виражена нечітко, переважно це межова розумова відсталість і дебільність різної тяжкості. Хворим властиві астенічні прояви і риси психічного інфантилізму: нестійкість уваги, підвищена втомлюваність, зниження працездатності та ін. Для синдрому Клайнфельтера характерна певна дисоціація між недорозвиненням інтелекту і незрілістю емоційно-вольової сфери.
Дослідження сперми виявляє зрілі форми сперматозоїдів тільки у дуже рідких випадках. Як правило, виявляється оліго або азооспермія. У пунктаті яєчка знаходять гіперплазію клітин Лейдига. Рівень фолітропіну значно підвищений.
Каріотипічна картина різноманітна: у більшості випадків виявляється класичний каріотип 47ХХY; зустрічаються і каріотипи 48ХХХY; 49ХХХХY, а також різні форми мозаїцизму: 47ХХY/46ХY; 47ХХY/46ХХ; 47ХХY/46ХY/46ХХ.
При трьох зайвих Х-хромосомах (49ХХХХY) симптомокомплекс настільки відрізняється від класичного синдрому Клайнфельтера, що деякі клініцисти виділяють його в окремий синдром - тетрасомію Х. При цьому синдромі відзначається низька маса тіла при народженні (в середньому 2600 г). Для зовнішнього вигляду характерне овальне обличчя, гіпертелоризм, косий розріз очей, епікант, косоокість, спинка носа трохи сплюснута, вдавлена, а кінчик носа піднятий. Рот великий, чітко окреслений, іноді наближається до трикутної форми. Вушні раковини великі, недорозвинені, розташовані нижче звичайного. Шия коротка, широка. З боку кістково-м'язової системи виявляються знижена рухомість у ліктьових суглобах, викривлення шийки стегна. Різко виражений гіпогонадизм, за допомогою біопсії знаходять ті ж зміни, що й у хворих з каріотипом 47ХХY. Розумова відсталість при тетрасомії ХY зустрічається у всіх випадках і відповідає глибокій дебільності або імбецильності.
Лікування синдрому Клайнфельтера головним чином гормональне. Його краще розпочинати з 10-12 років, терапія препаратами чоловічих статевих гормонів поліпшує фізичний стан. Призначають 1% чи 5% розчин тестостерону пропіонату. Виражену гінекомастію лікують хірургічним шляхом. У разы неглибокого зниження інтелекту застосовують психостимулятори і нейрометаболічні препарати.
Для стимуляції росту волосся на обличчі використовують розтирання і мазі, які містять андрогени.
Якщо каріотип батьків нормальний, ризик повторного народження дитини з синдромом Клайнфельтера не перевищує 1 %.
