-
Альбінізм
- Алкаптонурія
-
Галактоземія
-
Сімейна гіперхолестеринемія
-
Хвороба Тея — Сакса
-
Хвороба Німана – Піка
-
Хвороба Гоше
-
Синдром Леш — Ніхана
-
Синдром Дабіна
— Джонсона
-
Синдром Кріглера —
Найяра
-
Хвороба Вільсона — Коновалова
Тема 3. СПАДКОВІ ХВОРОБИ ОБМІНУ. ПРИНЦИПИ ЛІКУВАННЯ СПАДКОВИХ ХВОРОБ, РЕАБІЛІТАЦІЇ І СОЦІАЛОНОЇ АДАПТАЦІЇ.
Спадкові хвороби обміну речовин (СХО) — найчисленніша група аутосомно-рецесивних захворювань.
Переважна частина спадкових метаболічних розладів пов’язана з мутацією генів, що кодують ферменти (ферментопатії або ензимопатії). Класичним прикладом може бути група захворювань, обумовлених порушеннями обміну амінокислот.
Спадкові хвороби обміну можуть бути пов’язані також з порушенням структури клітинних рецепторів і каналів, транспортних білків, імунного захисту, системи виділення кінцевих продуктів метаболізму. У разі ферментопатій серед гетерозигот синтезується до 50 % ферменту і цього достатньо для забезпечення функції, тому більшість ферментопатій успадковується як рецесивні захворювання (аутосомно-рецесивні або зчеплені з Х-хромосомою). СХО пов’язані з порушенням будови структурних білків, можуть бути рецесивними і домінантними.
Існують різноманітні класифікації СХО — за типом успадкування, за характером метаболічних порушень, за клінічними проявами та ін., але жодна з них не є вичерпною.
За принципом провідних порушень обміну речовин виділяють такі типи:
- Порушення обміну амінокислот (аміноацидурії) — фенілкетонурія, гомоцистенурія, альбінізм, алкаптонурія та ін.
- Порушення обміну вуглеводів — галактоземія, глікогенози та ін.
- Порушення обміну ліпідів — ліпідози плазматичні (сімейна гіперхолестеринемія) і ліпідози клітинні — гангліозидози (хвороба Тея — Сакса), цереброзидози (хвороба Гоше) та ін.
- Порушення обміну пуринів і піримідинів — синдром Леш — Ніхана, окремі форми подагри та ін.
- Порушення обміну амонієвих сполук — дефіцит орнітинтранскарбамілази, гіпераргінінемія.
- Порушення обміну порфіринів — гостра інтермітуюча порфірія та ін.
- Порушення білірубінового обміну — синдроми Дубіна — Джонсона, Кріглера — Найяра та ін.
- Порушення обміну органічних кислот (органічні ацидемії) — пропіон, метилмалонова, ізовалеріанова ацидемії.
- Порушення обміну металів — хвороба Вільсона — Коновалова (обмін міді), гемохроматоз (обмін заліза) та ін.
- Порушення синтезу гормонів — гіпотиреоз, природжена гіперплазія надниркових залоз.
- Спадкові хвороби обміну сполучної тканини — мукополісахаридози, хвороба Марфана, синдром Елерса — Данло та ін.
- Порушення транспорту хлоридів — муковісцидоз.
- СХО транспортних систем нирок (тубулопатії) — цистинурія, вітамін-Д-резистентний рахіт та ін.
- Спадкові гемоглобінопатії.
- Лізосомні хвороби накопичення — мукополісахаридози, сфінголіпідози.
- Пероксисомні хвороби — хвороба Цельвегера.
- Мітохондріальні хвороби.
- Спадкові імунодефіцитні стани (СХО лейкоцитів і лімфоцитів).
- СХО еритрону — гемолітичні анемії, недостатність глюкозо-6-фосфатдегідрогенази та ін.
- СХО шлунково-кишкового тракту — синдром мальабсорбції при недостатності дисахаридаз та ін.
За клінічними проявами СХО можуть бути підрозділені таким чином:
· нейро-м’язові;
· ендокринопатії;
· печінкові;
· сполучної тканини;
· кишкові (синдром порушеного кишкового всмоктування);
· еритроцитарні (гемоглобінопатії);
· імунодефіцити;
· репарації ДНК;
· лізосомні — хвороби накопичення;
· мітохондріальні;
· пероксисомні.
Діагностика СХО за клінічними ознаками складна, оскільки клінічна картина спадкових хвороб обміну поліморфна і перекривається. Точний діагноз СХО можна встановити за допомогою лабораторних методів (скринінгові біохімічні з подальшим уточненням діагнозу). Однак при різних спадкових порушеннях метаболізму є загальні симптоми, які дозволяють запідозрити цю групу захворювань.
Альбінізм
Альбіні́зм (від лат. albus— білий) — уроджена відсутність пігменту шкіри, волосяного покриву, райдужки ока (рис.9).
|
Р |
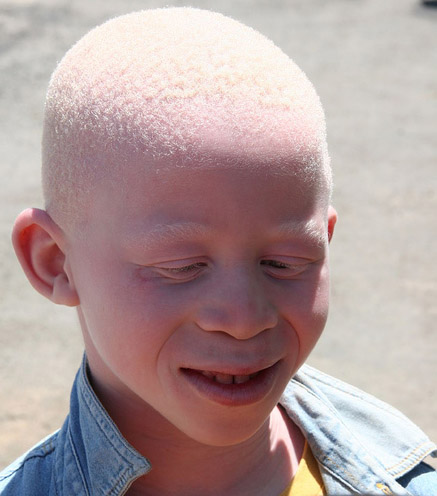 ис.
9. – Зовнішній вигляд хворого на
альбінізм
ис.
9. – Зовнішній вигляд хворого на
альбінізмАльбінізм може спостерігатися тільки у тих осіб, обоє батьків яких мають відповідний ген, при цьому ймовірність народження дитини-альбіноса при кожній вагітності складає 25%.
Частота появи осіб з альбінізмом варіюється залежно від регіону:
У Європі та Північній Америці вона становить від 1 на 17000 до 1 на 20000 дітонароджень. У деяких частинах Тихого океану цей показник дорівнює 1 на 700.
Серед деяких корінних народів Південної Америки - від 1 на 70 до 1 на 125.
У країнах Африки на південь від Сахари відзначається частота від 1 на 5000 до 1 на 15000, причому для окремих груп населення - від 1 на 1000 до 1 на 1500.
Важливим є те, що деякими дослідженнями щодо частоти альбінізму часто не вистачає об'єктивності в їх методології або ж вони є неповними, зводячи у більшості випадків ці оцінки до найбільш оптимальних припущень.
Етіологія
Вважається, що причиною захворювання є відсутність (або блокада) ферменту тирозинази, необхідної для нормального синтезу меланіну - особливої речовини, від якої залежить забарвлення тканин. Отже, білий колір альбіносів — не забарвлення, а його відсутність.
Види альбінізму:
тотальний
неповний
частковий альбінізм
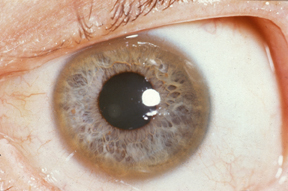
Тотальний альбінізм

Рис.10. - Очі людини, хворої на тотальний альбінізм (OCA1)
Успадковується аутосомно-рецесивно із середньою частотою 1: 10000-20000. Припускають, що носії мутантного гена становлять 1,5% усього населення світу. Депігментація шкіри і придатків спостерігається з народження, супроводжується сухістю шкіри, порушенням потовиділення, іноді гіпо-, або гіпертрихозом, особливо на відкритих ділянках. У хворих легко виникають сонячні опіки, актенічний хейліт. Вони схильні до розвитку кератиту, телеангіоектазій. Характерними є горизонтальний ністагм і виражена світлобоязнь. Часто спостерігається косоокість, зниження гостроти зору в результаті порушень рефракції, катаракти, можлива мікроофтальмія (рис.10),(рис.11). Нерідко спостерігається безпліддя, імунодефіцит (звідси періодичні інфекційні захворювання), вади розвитку, скорочення тривалості життя, олігофренія.

Рис. 11 - Очне дно у людини з тотальним альбінізмом (а) і у здорової людини (б)
Неповний альбінізм (альбіноідизм)
На відміну від попередньої форми, успадковується аутосомно-домінантно, у деяких випадках — рецесивно. Спостерігається зниження активності тирозинази, але не блокада її синтезу. Також характерною є гіпопігментація шкіри, волосся, райдужної оболонки, іноді фотофобія (несприйняття світла). Інших дефектів та аномалій немає.
Частковий альбінізм (піебалдизм)
Успадковується аутосомно-домінантно. Прояви виявляються при народженні. Характеризується появою ділянок ахромії на шкірі живота, нижніх кінцівок, пасмами сивого волосся. Депігментовані плями неправильної форми з різкими межами, на їх поверхні є дрібні темно-коричневі плями. Навколо ахромічних плям шкіра може бути пігментованою. Ураження інших органів, як правило, не буває. Частковий альбінізм є одним із проявів синдромів Чедіака-Хігасі, Клейна-Варденбурга, Тітце, Менде, Херманскі-Пудлака, Крос-МакКюзіка-Бріна.
Альбінізм очей
1.Нетлшіпа-Фолз.
Симптоми: депігментація очного дна з виступаючими судинами, ністагм, фотофобія, зниження гостроти зору, тремор голови, нормальна пігментація шкіри, мозаїчна картина депігментації очного дна в гетерозиготних носіях, макромеланосоми у електронній мікроскопії.
2.Форсіуса-Еріксона (хвороба Аландських островів).
Симптоми: депігментація очного дна, гіпоплазія зорової ямки, виражене зниження зору, ністагм, міопія, астигматизм, колірна сліпота, макромеланосоми при електронній мікроскопії відсутні.
3.Тип 3.
Симптоми: порушення зору, просвічення райдужної оболонки, вроджений ністагм, фотофобія, депігментоване очне дно, гіперплазія зорової ямки, косоокість.
Шкірно-очний альбінізм (ксантизм)
- Тирозиназа-негативний (жовтий альбінізм): дитина народжується блідою, потім поступово з'являється жовта пігментація шкіри і волосся, виражена очна патологія.
- Тирозиназа-позитивний: альбінізм, ністагм, зниження зору.
- TYRP1 (мутація гена тірозиназазалежного білка 1; ймовірно, алельних 2 типу): Неповний альбінізм, ністагм, присутній пігмент у сітківці, косоокість.
- Неповний (альбіноідизм): недостатня пігментація шкіри, волосся, точкові ділянки депігментації очного дна і райдужок, відсутність ністагму, фотофобія і порушення зору.
- З мінімальною пігментацією: відсутність пігментації шкіри і волосся після народженні, блакитні райдужні оболонки; пігментація відбувається протягом першого десятиріччя.
- Рудий тип (мутація гена TYRP1): спостерігають у афропоїдів, характерне мідно-червоне забарвлення волосся і шкіри.
Лікування альбінізму
Ефективних методів лікування немає. Рекомендовано застосовувати фотозахисні засоби, декоративну косметику. Для додання шкірі жовтуватого відтінку у разі тотального і неповного альбінізму рекомендують призначати бета-каротин (90-180 мг на добу). Слід рекомендувати хворому уникати сонячних опромінень і застосовувати світлозахисні засоби під час виходу на вулицю. З метою профілактики передачі аномалії у спадщину необхідні медико-генетичні консультації.
Алкаптонурія
Алкаптонурія - спадкове захворювання, обумовлене випадінням функцій оксидази гомогентизиновой кислоти і характеризується розладом обміну тирозину і екскрецією з сечею великої кількості гомогентизинової кислоти.
Етіологія
Алкаптонурія виникає внаслідок мутації гена, що кодує синтез оксидази гомогентезинової кислоти. Дана патологія характеризується аутосомно-рецесивним типом успадкування. На алкаптонурію частіше хворіють чоловіки. Ген оксидази гомогетинзинової кислоти людини (HGD) локалізований на довгому плечі 3 хромосоми людини (3q 21-23).
Патогенез
За нормальних умов гомогентизинова кислота - проміжний продукт розпаду тирозину і фенілаланіну - перетворюється у малеілацетооцтову кислоту, з якої у кінцевому результаті утворюються фумарова і ацетооцтова кислоти, що вступають в інші біохімічні цикли. Через дефект ферменту цей процес гальмується, і залишається в надлишку гомогентизинова кислота перетворюється поліфенолоксидазою у хінонову поліфенолову (алкаптон або бензохінонацетат), яка і виводиться нирками. Алкаптон не повністю екскретується сечею і починає відкладатися у хрящовій і інший сполучній тканині, обумовлюючи їх потемніння і підвищену крихкість. Найчастіше вперш за все з'являються пігментація склер і вушних хрящів.
Клінічні ознаки
Рання ознака алкаптонуріі - виділення у дитини сечі, яка швидко темніє при стоянні на повітрі, підігріванні, підлужнюванні. У подальшому може приєднатися сечокам'яна хвороба, яка ускладнюється пієлонефритом. Ознаки ураження опорно-рухового апарату з'являються зазвичай після 30 років. Характерне переважне ураження великих суглобів нижніх кінцівок: колінних, кульшових. Рідше до процесу залучаються плечові суглоби. Зміни характеризуються вторинним остеоартрозом. Відзначаються болі механічного характеру, часто виникає синовіїт, резистентний до лікування. Кількість запальних клітин у синовіальній рідині невелика. У багатьох пацієнтів відзначається швидке прогресування деструктивних змін хряща суглобів. Іноді термін, що проходить від дебюту суглобового синдрому до розвитку виражених змін, які потребують ендопротезування суглобів, може становити 2-3 роки. Нерідко відзначається надлишкове відкладення гомогентезинової кислоти у зв'язках, сухожилках і їх оболонках, що призводить у ряді випадків до розвитку локальних запальних змін і кальцифікації. Часто вражається хребет. Основні симптоми: біль і обмеження рухів переважно у поперековому відділі, рідше у грудному і шийному відділах хребта. На рентгенограмах виявляються зміни, характерні для розповсюдженого остеохондрозу, а також кальцифікація міжхребцевих дисків, що є характерною ознакою охронозу. Можливо як ізольоване ураження хребта, так і одночасне залучення великих суглобів. Клінічні ознаки ураження хребетного стовпа при алкаптонуріі можуть нагадувати анкілозуючий спондиліт. При обстеженні таких пацієнтів відзначається значне обмеження рухів хребта. При охронозі можуть виникати рентгенологічні зміни крижово-клубових суглобів (остеоартроз), частково подібні з такими при сакроілеїті. Ураження хрящової тканини вушних раковин зустрічається практично у всіх хворих на алкаптонурію в розгорнутій стадії хвороби. При цьому змінюється колір вушних раковин: він може варіювати від блакитного до сірого, забарвлення може бути як інтенсивним, так і злегка помітним. Змінюється також еластичність вушних раковин: при пальпації вони стають більш щільними і ригідними. Рідше змінюється колір шкіри в області носо-губних складок, пахвових западин, долонь. Ці зміни протікають безсимптомно. Дуже часто у хворих на алкаптонурію розвивається пігментація склер, що пов'язано з відкладанням у них депозитів гомогентезинової кислоти. Інтенсивність таких відкладень може бути різною. Ці зміни зазвичай не турбують пацієнтів, але є одними з ознак даного захворювання, яке має важливе діагностичне значення. У разі алкаптонуріі приблизно у 20% хворих розвиваються зміни аортального клапана (рідко - мітрального): кальцифікація стулок, фіброзного кільця, а також висхідного відділу аорти. Ці зміни можуть бути значними, призводити до істотних гемодинамічним порушень, які вимагають у ряді випадків оперативного лікування (протезування клапанів). Є дані про розвиток кальцинозу коронарних артерій. Калькульозний простатит часто виявляється при алкаптонуріі. Зазвичай він протікає мало або безсимптомно, виявляється при ультразвуковому або рентгенологічному дослідженні.
Діагностика
Найбільш інформативним для діагностики алкаптонуріі є метод кількісного визначення гомогентизинової кислоти і бензохінооцтової кислоти у сечі. Для цього використовується ферментативна спектрофотометрія або рідинна хроматографія. Більш простим, але менш точним способом виявлення даного захворювання є оцінка кольору сечі через 12-24 год після перебування її на повітрі. У цьому випадку відбувається окислення алкаптона, що призводить до зміни кольору сечі (стає бурою або чорною). Дані зміни відбуваються тільки за лужних значеннь рН сечі, тому при кислій реакції сечі необхідно її підлужнювання. У ряді випадків діагноз «алкаптонурія» може бути встановлений у разі виявлення характерної пігментації хряща у ході артроскопії, синовіальної оболонки під час мікроскопічного дослідження або клапанів серця в ході їх протезування. Важливо розмежувати генетичну алкаптонурію від алкаптонурії при гіповітамінозі С. Остання зникає після призначення адекватної дози аскорбінової кислоти. Слід відрізняти алкаптонурію від гематурії, гемоглобінурії, меланінуріі, порфірії.Лікування і профілактика
Радикального лікування немає, використовується симптоматична терапія і великі дози аскорбінової кислоти.
Генетична схильність
Це захворювання успадковується генетично за аутосомно-рецесивним типом. Батьки людини з аутосомно-рецесивним захворюванням є носіями по одній копії кожного з мутаційних генів, але зазвичай у них немає ознак захворювання.Галактоземія
Галактоземія - спадкове захворювання, в основі якого лежить порушення обміну речовин на шляху перетворення галактози в глюкозу (мутація структурного гена, відповідального за синтез ферменту галактозо-1-фосфатурідилтрансферази).
Етіологія і патогенез
Галактоза, що надходить з їжею у складі молочного цукру - лактози, піддається перетворенню, але реакція перетворення не закінчується через спадковий дефект ключового ферменту.
Галактоза і її похідна накопичуються в крові і тканинах, надаючи токсичну дію на центральну нервову систему, печінку і кришталик ока, що визначає клінічні прояви хвороби. Тип успадкування галактоземії аутосомно-рецесивний.
Клінічні ознаки
Захворювання проявляється в перші дні і тижні життя вираженою жовтяницею, збільшенням печінки, блюванням, відмовою від їжі, зниженням маси тіла, неврологічною симптоматикою (судоми, ністагм (мимовільний рух очних яблук), гіпотонією м'язів; в подальшому виявляється відставання у фізичному і нервово-психічному розвитку, розвивається розумова відсталість, виникає катаракта. Тяжкість захворювання може значно варіювати, іноді єдиним проявом галактоземії бувають лише катаракта або непереносимість молока. Класична галактоземія часто носить життєвозагрожуючий характер. Один із варіантів хвороби - форма Дюарте - протікає безсимптомно, хоча відзначена схильність таких осіб до хронічних захворювань печінки.
При лабораторному дослідженні в крові визначається галактоза, вміст якої може досягати 0,8 г / л; спеціальними методами (хроматографія) вдається виявити галактозу у сечі. Активність ферментів в еритроцитах різко знижена або не визначається, вміст ферментів збільшено в 10-20 разів у порівнянні з нормою. При наявності жовтяниці наростає вміст як прямого (диглюкороніду), так і непрямого (вільного) білірубіну. Характерні і інші біохімічні ознаки ураження печінки (гіпопротеїнемія, гіпоальбумінемія, позитивні проби на порушення колоїдостійкості білків). Значно знижується опірність по відношенню до інфекції. Можливий прояв і геморагічного діатезу через зменшення протеїносинтетичної функції печінки і зменшення числа тромбоцитів - петехії.
Діагностика
В даний час проводиться масове обстеження новонароджених на галактоземії. Визначається рівень тотальної (загальної) галактози в плямах висушеної крові флуоресцентним методом. При виявленні підвищеного рівня загальної галактози проводиться визначення рівня активності ферменту АЛТ.
Важкі форми закінчуються летально у перші тижні життя, при затяжному перебігу на перший план можуть виступати явища хронічної недостатності печінки або ураження центральної нервової системи.
Лікування і профілактика
У разі підтвердження діагнозу необхідно перевести дитину на харчування із виключенням, головним чином, молока. Для цього розроблені спеціальні продукти: сояваль, Нутраміген, безлактозний енпіт. Рекомендуються замінне переливання крові, дробові гемотрансфузії, вливання плазми. З лікарських препаратів показано призначення оротата калію, АТФ, кокарбоксилази, комплекс вітамінів.
Показано високу ефективність раннього виявлення вагітних у сім'ях високого ризику і внутрішньоутробної профілактики, яка складається у виключенні молока з дієти вагітних.
Облік сімей ризику дозволяє рано, тобто ще в доклінічній стадії, піддати спеціальному обстеженню новонародженого і у разі позитивних результатів перевести його на безлактозне вигодовування. Для раннього виявлення запропоновані також спеціальні скринінг-програми масового обстеження новонароджених.
З віком спостерігається ослаблення цього специфічного порушення обміну.
Сімейна гіперхолестеринемія
Сімейна гіперхолестеринемія — генетичне захворювання, яке супроводжується високим рівнем холестерину, особливо холестерину ліпопротеїнів високої щільності та раннім розвитком серцево-судинних захворювань.
Клінічні ознаки
Високий рівень холестерину в крові сам по собі не викликає ніяких симптомів. Людина навіть не здогадується про свою проблему, поки не здасть аналізи крові. Симптоми цього захворювання можуть бути наступними:
• Високий рівень холестерину в крові пацієнта.
• Холестеринові відкладення на колінах, ліктях і сідницях - ксантоми.
• Біль у грудях, викликана звуженням коронарних судин - стенокардія.
• Інфаркти, які трапляються у відносно ранньому віці.
Патогенез.
Холестерин доставляється до клітин з током крові. Молекули ЛПНЩ прикріплюються до особливих клітинних рецепторів, як би створені для них (за принципом «ключ до замка»). Завдяки наявності цих рецепторів холестерин проникає до клітини і виконує свої природні функції. Ген в 19-й хромосомі (LDLR) - кодує ці клітинні рецептори. При сімейної гіперхолестеринемії у хворих успадковується мутація LDLR-гена, яка порушує розвиток холестеринових рецепторів, їх кількість і структуру. Це означає, що ЛПНЩ недостатньо добре засвоюються клітинами, залишаючись у вільному вигляді в кровотоці. Високий рівень ЛПНЩ в крові призводить до розвитку атеросклерозу - основної причини інфарктів та інших важких захворювань.
Успадкування
Сімейна гіперхолестеринемія - це так зване аутосомно-домінантне захворювання. У великій кількості випадків дефектний ген успадковується від одного носія- в рідкісних випадках носіями є обоє батьків.
1. Дефектний ген у одного з батьків. Якщо один з батьків є носієм одного нормального і одного дефектного гена у парі, то кожна дитина має 50% ймовірність успадкування одного гена, що мутує. Ризик розвитку захворювань коронарних судин в ранньому віці залежить від віку і статі хворої дитини:
• У 50% чоловіків з цим захворюванням хвороби коронарних артерій розвиваються до того, як вони досягають 50-річного віку.
• Всі 100% чоловіків, народжених від носія сімейної гіперліпідемії, будуть страждати коронарними захворювання до 70 років.
• Приблизно у 85% таких чоловіків може статися інфаркт міокарда до досягнення ними 60-річного віку.
• У 12% жінок, які успадкували цю мутацію, виникнуть захворювання коронарних артерій до 50-річного віку. Близько 74% з них будуть страждати коронарними захворюваннями до 70 років.
2. Дефектний ген у обох батьків. У цьому випадку прогноз більш несприятливий - кожна четверта дитина (25%) успадковує відразу два дефектних гена у парі. При цьому у дитини розвиваються важкі коронарні захворювання у перші десятиріччя життя, навіть в дитячому віці. Ця форма сімейної гіперхолестеринемії стійка до лікування. Незважаючи на всі заходи, ризик серцевого нападу залишається дуже високим. У хворих може спостерігатися інтенсивне відкладення надлишкового холестерину під шкірою - численні ксантоми на колінах, ліктях, сідницях.
Діагностика
Діагноз сімейної гіперхолестеринемії встановлюється за результатами багатьох тестів, серед яких:
• Фізичний огляд.
• Аналізи крові на холестерин.
• Дослідження серця (стрес-тест).
• Генетичний аналіз для підтвердження.
Лікування
Оскільки це захворювання спадкове, то повного лікування від нього немає. Терапія направлена на зниження ризику захворювань коронарних артерій і інфаркту. Рекомендації включають:
Зміна раціону. Хворим настійно рекомендується зменшити споживання продуктів, які містять холестерин і насичені жири. Також корисно споживати більше клітковини - свіжі фрукти, овочі. Після 3-4 місяців такої дієти лікар проводить повторні аналізи, щоб визначитися з подальшими заходами. У будь-якому випадку, дієта - це найважливіша складова лікування.
Рослинні стероли і станоли. Ці речовини за структурою схожі на холестерин, але вони не засвоюються клітинами організму. Дослідження показали, що вживання стеролов і станолів призводить до зниження рівня холестерину в крові. Джерелами цих речовин є кукурудза, рис, горіхи, рослинні олії.
Вправи. Регулярні фізичні вправи сприяють зниженню рівня холестерину, що підтверджується численними дослідженнями.
Контроль маси тіла. Ожиріння - це дуже суттєвий фактор ризику. Підтримка нормального індексу маси тіла (ІМТ) - це одна з основних цілей терапії. Крім зниження ризику атеросклерозу, боротьба з ожирінням допоможе зменшити ймовірність цукрового діабету та інших захворювань.
Відмова від паління. Токсичні компоненти тютюну ініціюють первинне ушкодження судин, з яких починається атеросклероз і поступова закупорка артерій.
Медикаментозна терапія. У дуже невеликого відсотка хворих на сімейну гіперліпідемію виходить знизити свій холестерин без ліків. У переважній більшості випадків хворим потрібен постійний прийом спеціальних препаратів, таких як статини (аторвастатин, симвастатин, ловастатин).
Додаткові методи лікування: аферез ЛПНЩ, що представляє собою очищення крові від ліпопротеїнів через спеціальний апарат.
Хвороба Тея — Сакса
Хвороба Тея-Сакса (ХТС) (також відома як GM2 гангліоліпідоз або дефіцит гексозамінідази або рання дитяча амавротична ідіотія) — аутосомно-рецесивне генетичне захворювання, яке спричинює прогресуюче погіршення розумових і фізичних здібностей дитини. Перші ознаки захворювання зазвичай проявляються у віці приблизно 6 місяців. Розлад, як правило призводить до смерті хворої особи у віці близько 4 років.
Захворювання спричинює генетичний дефект конкретного гена. Якщо народжена дитина уражена ХТС, то це означає, що вона успадкувала по одній копії дефектного гена від кожного з батьків. Захворювання проявляється тоді, коли у нервових клітинах мозку накопичується небезпечна кількість гангліозидів, що в результаті призводить до передчасної смерті цих клітин. На сьогодні не існує жодних ефективних ліків чи інших методів лікування цієї хвороби. ХТС зустрічається досить рідко, у порівнянні з іншими рецесивними захворюваннями, такими як, наприклад, муковісцидоз (кістозний фіброз) і серповидно-клітинна анемія — які є набагато поширенішими.
Хвороба названа на честь британського офтальмолога Уоррена Тея, який першим описав червону пляму на сітківці ока у 1881 році, і американського невролога Бернарда Сакса.
Дослідження захворювання, які проводилися наприкінці XX століття показали, що хвороба Тея-Сакса обумовлена мутаціями гену HEXA, який знаходиться на 15 хромосомі. На сьогодні вже виявлена велика кількість мутацій HEXA, а нові дослідження надають інформацію про нові мутації. Ці мутації дуже часто зустрічаються в декількох популяцій.
Класифікація та клінічні прояви
Хвороба Тея-Сакса класифікується за різними формами, в залежності від часу виникнення неврологічних симптомів. Форма захворювання відображає варіант мутації.
Дитяча форма хвороби Тея-Сакса
Після народження впродовж перших шести місяців діти розвиваються нормально. Але, після того як нервові клітини накопичують гангліозиди і, таким чином розтягуються, спостерігається невпинне погіршення розумових і фізичних здібностей хворого. Дитина стає сліпою, глухою, і не може ковтати. М'язи починають атрофуватися в наслідок чого настає параліч. Смерть зазвичай наступає у віці до чотирьох років.
Підліткова форма хвороби Тея-Сакса
Ця форма захворювання зустрічається вкрай рідко і зазвичай проявляє себе у дітей у віці від 2 до 10 років. У них розвиваються когнітивно-моторні проблеми, дизартрія, дисфагія, атаксія, виникає спастичність. Пацієнти з підлітковою формою ХТС зазвичай помирають у віці від 5 до 15 років.
Доросла форма хвороби Тея-Сакса
Рідкісна форма розладу, відомого як доросла форма хвороби Тея-Сакса або пізня форма хвороби Тея-Сакса виникає у пацієнтів у віці від 20 до 30 років, часто неправильно діагностується, і, як правило, не має летальних наслідків. Вона характеризується порушенням ходи та прогресуючим погіршенням неврологічних функцій. До симптомів даної форми, яка виникає у підлітковому або ранньому дорослому віці, відносять: проблеми із мовленням та ковтанням, хиткість ходи, спастичність, зниження когнітивних навичок, виникнення психічних захворювань, зокрема шизофренії у вигляді психозу.
Доросла форма ХТС
ХТС, яка виникє у підлітковому чи дорослому віці часто діагностують як неврологічні розлади, наприклад як атаксію Фридрейха. Особи, уражені ХТС у дорослому віці часто пересуваються за допомогою інвалідного крісла, проте багато з них живуть майже повноцінними життям, але лише в тому випадку, якщо пристосуються до фізичних та психіатричних ускладнень (які можна контролювати за допомогою медичних препаратів).
Патофізіологія
Хвороба Тея-Сакса виникає внаслідок недостатньої активності ферменту гексозамінідази А, який каталізує біодеградацію певного классу жирних кислот відомих як гангліозиди. Гексозамінідаза А є життєво необхідним гідролітичним ферментом, який знаходиться в лізосомах та руйнує ліпіди. Коли гексозамінідаза А перестає функціонувати належним чином, ліпіди накопичуються в головному мозку і перешкоджають нормальним біологічним процесам. Гангліозиди виробляються та біодеградують швидко на самому початку життя, в той час, як розвивається мозок. Пацієнти і носії хвороби Тея-Сакса, можуть бути визначені шляхом біохімічного аналізу крові, який визначає активність гексозамінідази А.
Для гідролізу GM2-гангліозид необхідними є три білки. Два з них є субодиницями гексозамінідази А, а третій — невеликий гліколіпідний транспортний білок, GM2 білок-активатор (GM2A), який виступає як субстрат для конкретного кофактора фермента. Дефіцит будь-якого з цих білків призводить до накопичення гангліозидів, головним чином у лізосомах нервових клітин. Хвороба Тея-Сакса (разом з GM2 гангліозидозом та хворобою Сандхоффа) виникає через генетичні мутації успадковані від обох батьків, які або деактивують, або гальмують процес розщеплення цих речовин. Більшість мутацій ХТС, як відомо, не впливають на функціональні елементи білка. Замість цього, вони спричиняють неправильне накопичення або зберігання ферменту, у зв'язку з чим внутрішньоклітинне транспортування стає неможливим.
ХТС — це аутосомно-рецесивне генетичне захворювання. Це означає, що у тому випадку, якщо обидва батьки є носіями дефектного гену, то ризик того, що новонароджена дитина буде хворою становить 25%. Кожна людина є носієм двох копій кожного аутосомного гена, по одній успадкованій від кожного з батьків. Якщо обоє батьків – носії мутації, то відповідно до генетичних законів Менделя, ймовірність передачі хвороби дитині становить 25%. Як і всі генетичні захворювання, ХТС може виникнути у будь-якому поколінні, при цьому не важливо, коли вперше виникла мутація. Хоча мутації, які викликають ХТС зустрічаються доволі рідко.
Аутосомно-рецесивні захворювання виникають, якщо дитина успадковує дві копії дефектного аутосомного гена, тобто коли жодна копія не може брати участі в процесі транскрипції або експресії як функціональний продукт для утворення ферменту.
Діагностика
Удосконалення розроблених методів тестування дозволило невропатологам набагато точніше діагностувати хворобу Тея-Сакса та інші неврологічні захворювання. У пацієнтів з даним захворюванням наявна «вишнева пляма», яку легко виявити лікареві за допомогою офтальмоскопа на сітківці ока. Ця пляма є ділянкою сітківки, яка збільшується через накопичення гангліозидів у навколишніх гангліозних клітинах сітківки (вони є нейронами центральної нервової системи). Таким чином, тільки вишнева плями макули є тією частиною сітківки, яка забезпечує нормальний зір. Мікроскопічний аналіз нейронів показує, що вони є розтягнутими (навантажені гангліозидами) у зв'язку із надлишковим накопиченням гангліозидів. Без використання молекулярних методів діагностики, тільки вишнева плями макули є характерною рисою та ознакою для діагностики всіх гангліозидозів.
На відміну від деяких інших лізосомних хвороб накопичення (наприклад, хвороби Гоше, Німана-Піка, Сандхоффа), гепатоспленомегалія не є характерною ознакою хвороби Тея-Сакса.
Скринінг
Скринінг ХТС здійснюють двома можливими шляхами:
Тест на визначення носія. Під час його проведення виявляють чи людина неуражена хворобою є носієм однієї копії мутації. Багато людей, охочих пройти тест на визначення носія, є парами з груп ризику, які планують створити сім'ю. Деякі особи та подружні пари хочуть пройти скринінг, оскільки вони знають про наявність генетичного захворювання у прабатьків або членів їх родини.
Пренатальне тестування допомагає визначити, чи успадкував плід дві дефектні копії гена, по одній від кожного з батьків. При використанні даної діагностики, як правило, кількість інформації, щодо сімейної історії і мутації (які відомі точно) є більшою. Пренатальне тестування для ХТС, зазвичай, виконують, якщо обоє батьків не можуть бути виключеними як можливі носії. У деяких випадках, статус матері, може бути відомий, а батька або невідомий або недоступний для тестування. Цей тест може бути виконаний через аналіз активності ферменту HEXA у ембріональних клітинах отриманих шляхом біопсії хоріона або амніоцентезу. Якщо конкретна мутація була виявлена у обох батьків, то можна зробити точніше дослідження за допомогою технологій для аналізу мутацій, а саме ПЛР (полімеразна ланцюгова реакція, PCR).
Доступними є два технічних підходи до тестування мутацій Тея-Сакса. Першим підходом є випробування ферментативної активності, коли тестується фенотип на молекулярному рівні шляхом вимірювання рівня активності ферментів, у той час як аналіз мутацій (другий підхід) тестує безпосередньо генотип, шукаючи відомі генетичні маркери. Як і у всіх медико-біологічних дослідженнях, для обох підходів характерними є хибно позитивні та хибно негативні результати. Два методи використовуються паралельно, оскільки випробування ферментативної активності може виявитися зміненим, у випадку всіх мутації, проте з деякими непереконливими результатами, у той час як аналіз мутацій надає достовірні результати, але тільки для відомих мутацій. Сімейна історія може бути використана для вибору ефективнішого напрямку тестування.
Хвороба Німана – Піка
Хвороба Німана-Піка (ХНП) належить до летальних спадкових порушень обміну речовин, які входять у велику групу лізосомних хвороб накопичення.
Клінічні ознаки
Основні ознаки захворювання пов’язані з тими органами, в яких накопичуються шкідливі для організму речовини. Збільшення печінки і селезінки може викликати зниження апетиту, здуття і біль у животі. Наслідком спленомегалії може бути тромбоцитопенія.
Накопичення сфінгомієліну в органах центральної нервової системи (у тому числі у мозочку) призводить до:
- розладу координації довільних рухів (атаксії);
- невиразного мовлення (дизартрії);
- порушення ковтання (дисфагії).
Дисфункція базальних гангліїв призводить до дистонії. А пошкодження верхніх ділянок стовбура головного мозку спричиняє порушення швидких рухів очей (параліч м’язів ока). Найчастіше, захворювання впливає на кору головного мозку та підкіркові структури, що веде до поступової втрати інтелектуальних здібностей, викликаючи деменцію та епілепсію.
Відомі також випадки виникнення у хворих таких розладів як катаплексія (втрата м’язового тонусу і, навіть, втрата свідомості, які можуть виникати на фоні сильних емоційних реакцій), спричинена раптовим, безпричинним сміхом. Часто у людей, уражених ХНП відбувається так звана інверсія сну, коли вони хочуть спати вдень і страждають безсонням вночі.
Причини
Два типи хвороби Німана-Піка, А і В виникають внаслідок мутацій гену SMPD1, в той час як хворобу Німана-Піка типу С, спричинюють мутації, які відбуваються в генах NPC1 і NPC2.
Хвороба Німана-Піка успадковується за аутосомно-рецесивним типом тобто для того, щоб дитина успадкувала це захворювання обидві копії чи алелі гена, успадковані від батьків - повинні бути мутовані (змінені таким чином, що функції гена порушуються, на відміну від поліморфізму, в якому нуклеотидна послідовність змінюється, не викликаючи при цьому жодних функціональних порушень). При цьому, батьки хворої дитини, найчастіше є носіями захворювання, і жодних ознак чи симптомів хвороби у них не проявляється. Якщо обидва батьки – носії ХНП, то ймовірність того, що дитина народиться хворою становить 25%. Саме тому, для тих сімей, де відомі випадки захворювання необхідно здійснити генетичне тестування та звернутися за генетичною консультацією.
Класифікація (1961р.)
- хвороба Німана-Піка, тип А: класична інфантильна;
- хвороба Німана-Піка, тип В: вісцеральна;
- хвороба Німана-Піка, тип С: негостра підліткова;
- хвороба Німана-Піка, тип D: ново-шотландська.
На сьогодні, коли зрозуміла генетична природа захворювання, розлад класифікується наступним чином:
- хвороба Німана-Піка, пов’язана з геном SMPD1, яка включає в себе типи А і В;
- хвороба Німана-Піка, типу C, який включає в себе типи C1 і C2. (Тип D виникає в результаті мутації того ж гена, що й тип C1).
Патофізіологія
Захворювання Німана-Піка типів А, В та С – це генетичні захворювання, які відносяться до сфінголіпідозів чи ліпідозів (підгрупи лізосомних хвороб накопичення), при яких ліпіди, накопичуються у селезінці, печінці, легенях, кістковому і головному мозку. При (класичному, інфантильному) типі А захворювання, яке спричиняється міссенс мутацією, виникає дефіцит сфінгомієлінази.
Сфінгомієлін – це один з компонентів клітинної мембрани у тому числі мембранних органел. Дефіцит ферменту сфінгомієлази порушує процес розщеплення ліпідів, внаслідок чого він накопичується у макрофагах (моноцитах та фагоцитах). Ці клітини, іноді збільшуються до 90 мікрометрів у діаметрі. Крім того, накопичення сфінгомієліну та холестеролу призводить до розтягування лізосом. Під час гістологічного дослідження препаратів, отриманих від пацієнта з хворобою Німана-Піка, у макрофагах кісткового мозку спостерігається надлишок ліпідів та присутність так званих «блакитних гістіоцитів». У цитоплазмі утворюються численні невеликі вакуолі однакової форми і розміру, які створюють ефект піни у цитоплазмі.
Лікування
Можливості лікування хвороби Німанн-Піка в основному є обмеженими і переважно застосовується підтримуюча та симптоматична терапія. Варто зазначити, що операції з трансплантації органів, поки що не дуже успішні. В майбутньому вчені розраховують на те, що для лікування цього захворювання можна буде застосовувати технології ферментної заміни та генну терапію. При захворюванні на тип В, може бути здійснена трансплантація кісткового мозку.
Хвороба Гоше
Хвороба Гоше - спадкове захворювання накопичувального характеру, яке є одним з найпоширеніших серед лізосомних патологій. Хвороба Гоше розвивається внаслідок недостатності глюкоцереброзидази, що стає результатом накопичення глюкоцереброзида у тканинах і деяких органах.
Причини
На генетичному рівні відбуваються мутації в генах, які відповідають за вироблення ферменту глюкоцереброзидази. Цей ген з аномаліями локалізується на 1-й хромосомі. Дані мутації стають причиною низької активності ферменту. Таким чином, відбувається накопичення глюкоцереброзиди у макрофагах. Мезенхімальні клітини, що отримали назву клітин Гоше, поступово розростаються і стають гіпертрофованими. Так як у цих клітинах відбуваються видозміни, а вони знаходяться у селезінці, нирках, печінці, легенях, головному і кістковому мозку, то вони, в свою чергу, деформують ці органи і порушують їх нормальне функціонування. Хвороба Гоше належить до аутосомно-рецесивних захворювань. Тому будь-яка людина може успадкувати мутацію даного ферменту з усіма особливостями в однаковому співвідношенні, як від батька, і від матері. Таким чином, ступінь захворювання та її вираженість будуть залежати від ураження генів. Теоретично, кожна людина може успадкувати ген глюкоцереброзиди з ураженнями або абсолютно здоровий. В результаті успадкування гена з аномаліями, відбувається мутація даного ферменту, але це ще не говорить про хворобу. А от коли дитина отримує обидва уражених гена, то тоді виставляють діагноз хвороба Гоше. При успадкуванні одного ураженого гена, дитина вважається тільки носієм захворювання. Таким чином, якщо носії захворювання обидва батьки, дитина може народитися з хворобою Гоше у 25% випадках, дитина-носій - у 50% і здоровий - у 25%. Частота даної спадкової патології серед етнічних рас складає 1:50000 але набагато частіше вона виявляється серед євреїв-ашкеназі. Хвороба Гоше називається ще хворобою накопичення через недостатність ферменту, який повинен виводити шкідливі продукти обміну з організму, а не накопичувати їх. В результаті цього, ці речовини збираються в макрофагах деяких органів і руйнують їх.
Клінічні ознаки
Клінічна картина захворювання характеризується трьома типами. При першому типі хвороби Гоше нервова система не бере участь у накопиченні ліпідів, тому його відносять до гематологічного перебігу хвороби. В першу чергу, відбувається збільшення багатьох паренхіматозних органів, на першому місці серед яких знаходиться селезінка, а потім спостерігаються візуальні деформації з боку кісток. Набагато частіше цей тип захворювання прогресує серед дорослих.
Дитячий, або другий тип хвороби Гоше характеризується гострою формою неврології та діагностується частіше до шестимісячного віку. В даному випадку дуже рано з'являються характерні неврологічні симптоми, при яких ушкоджуються стовбурові клітини мозку, що стає причиною летального результату у дітей. Третій тип хвороби Гоше - це ювенільна форма перебігу хвороби з підгострим нейровісцеральним типом. На початку патологічного процесу розвивається спленомегалія та розумова відсталість, яка призводить до ураження пірамідних і екстрапірамідних систем головного мозку. Хвороба Гоше має основні зміни у вигляді ураження кісткового мозку, селезінки, лімфатичних вузлів і печінки, у разі церебральної форми уражається ще й головний мозок. В паренхіматозних органах знаходиться великий вміст клітин Гоше, а при наявних ділянках некрозу, в корі головного мозку відбуваються дифузні зміни. Під час обстеження хворих відзначаються різко збільшені селезінка і печінка, а іноді і лімфатичні вузли. На тілі з'являються характерні для цього захворювання плями темно-жовтого кольору. Хвороба Гоше має характерний симптом у клініці захворювання у вигляді збільшення селезінки на самому початку розвивитку патології, а потім вже відбувається збільшення в розмірах і печінки. Спостерігається певна пігментація на обличчі, кистях рук, склер та слизових оболонках. А накопичення ліпідів в кістковому мозку призводить до остеопорозу хребців, стегнових кісток, до спонтанних переломів, анемії, тромбоцитопенії та лейкопенії. Іноді у новонароджених зустрічається навіть злоякісна форма захворювання, що протікає у вигляді спастичного синдрому, косоокості, кашлюкоподобного кашлю. При гострій формі хвороби Гоше виявляється гепатоспленомегалія зі збільшеними вісцеральними лімфатичними вузлами. Відкриті частини тіла займає пігментація жовтувато-коричневого забарвлення, яка в подальшому стає бронзового кольору через відкладання певного пігменту під шкірою. Хворі, як правило, скаржаться на біль у кістках, характерні деформації і часті переломи кісток. У літньому віці добре простежуються виражені симптоми атероматоза судин одночасно з підвищеною холестеринемією. На останньому етапі хвороби Гоше дуже часто розвиваються тяжкі форми неврологічного розладу, які призводять до смертельного результату.
Діагностика
Сьогодні для діагностування хвороби Гоше використовуються різні методи. Одним з найбільш точних вважається аналіз крові, при якому визначають наявність ферменту і рівень глюкоцереброзидази у лейкоцитах. При аналізі ДНК, другому методі, розглядають на генетичному рівні мутації і недостатню кількість основного ферменту. Цей метод заснований на новітніх технологіях молекулярної біології. Головна перевага цієї діагностики хвороби Гоше полягає в тому, що вона дозволяє проводити обстеження на ранніх термінах вагітності. А це дозволяє на 90%, в майбутньому, визначити носія захворювання і навіть ступінь його тяжкості. При третьому способі діагностики досліджують кістковий мозок, що дає можливість визначити дисфункції його клітин. Негативна сторона такого обстеження полягає в тому, що воно дозволяє діагностувати тільки хворих, а виявити носіїв захворювання воно не може. Тому на сьогодні такий метод практично не застосовується. На жаль, іноді симптоми хвороби Гоше виявляються не завжди легкими. Тому саме правильна і своєчасна діагностика може продовжити хворому життя на багато років. При цьому пацієнт буде перебувати під наглядом фахівців (педіатрів, гематологів), не вдаючись до використання медикаментозного лікування і не побоюючись появи ускладнень. І, навпаки, якщо хворий, який своєчасно не пройшов кваліфіковану діагностику і не отримав медичну допомогу, може протягом усього життя мати симптоми хвороби, страждати від них і не здогадуватися, що у нього хвороба Гоше, відповідно, і не отримати правильного лікування.
Лікування
На даний момент у всьому світі величезна кількість хворих на хворобу Гоше постійно отримують ферментозамісну терапію у вигляді модифікованих форм бета-глюкоцереброзидази. До них відносяться церадаза (альглюцераза) або церезим (імиглюцераза в ін'єкціях). Ці препарати створені таким чином, що можуть спеціально впливати на білі кров'яні клітини (макрофаги), які знищують інфекції, для прискорення процесу розщеплення глюкоцереброзидів на глюкозу і кераміди. Гарний успіх лікування хвороби Гоше досягається у разі використанні церезима в початковій дозі 60 ОД на кг, яка застосовувалася через кожні два тижні. Після проходження курсу, зареєстровано, що саме це дозування значно знижує органомегалію, часто зменшує ускладнення гематологічної етіології, а також покращує життя хворих першим типом хвороби Гоше.
Тривале лікування хвороби Гоше цим препаратом повністю стабілізує патологічний процес, знижує виражені зміни у кістках і помітно покращує рівень життя хворих. Тому, чим раніше буде розпочато відповідна терапія, тим ефективнішими будуть результати.
Важливим моментом є профілактика хвороби Гоше, яка полягає у проведенні медико-генетичного консультування сімей та пренатальної діагностики. Це дає можливість визначити на ранніх етапах активність глюкоцереброзидази ще в оболонці плоду і кров'яних клітинах його пуповини.
Синдром Леш — Ніхана
Синдром Леша-Ніхана - спадкове захворювання, що характеризується збільшенням синтезу сечової кислоти (у дітей) викликане дефектом ферменту гіпоксантин-гуанинфосфорибозилтрансферази, який каталізує реутилізацію гуаніну і гіпоксантину - в результаті утворюється більша кількість ксантину і, отже, сечової кислоти.
Причини
Спадкова хвороба обміну речовин, обумовлена дефіцитом ферменту гіпоксантин-фосфорибозилтрансферази, що виявляється розумовою відсталістю, хореоатетоз, нападами агресивної поведінки з самоушкодженням, підвищеним вмістом сечової кислоти у сечі. Ген, що кодує гіпоксантин-фосфорибозилтрансферазу, розташований у X-хромосомі. Захворювання успадковується як моногенна рецесивна X-зчеплена ознака.
Патогенез
Ферментативний дефект призводить до порушення пуринового обміну і підвищеної продукції сечової кислоти. Гіперурикемія викликає дефіцит дофаміну у всіх підкіркових структурах, за винятком чорної субстанції, що, ймовірно, є наслідком порушення розгалуження терминалей дофамінергічних нейронів, у тому числі в стріатумі.
В наслідок чого розвивається гіперчутливість D1-рецепторів на стріарних нейронах, з якої можна частково пов'язати аутоагресивні дії.
Клінічні ознаки
Захворювання відзначається в осіб чоловічої статі. На першому році життя проявляється затримка психомоторного розвитку, в подальшому приєднуються спастичність і хореоатетоз. Характерною ознакою хвороби є аутоагресивні дії, які зазвичай розвиваються незабаром після того, як у дітей прорізуються зуби. Хворі обкушувають собі губи, нігті, пальці, передпліччя (аж до само ампутації), дряпають ніс і рот, пускають собі кров. Больова чутливість залишається збереженою. У зв'язку з цим хворі нерідко кричать від болю, яку самі ж собі заподіяли. Вони можуть також демонструвати агресію і по відношенню до інших людей, трощити все довкола. В деяких випадках стан покращується при прийомі леводопи і антагоніста опіатів налтрексону
Діагностика
Діагноз синдрому Леша-Ніхана ставиться за трьома основними клінічними елементами: підвищена продукція сечової кислоти, неврологічна дисфункція, когнітивні та поведінкові порушення. Досить складно поставити діагноз на ранній стадії, коли ці три ознаки не так очевидні. Підозри можуть виникнути через затримку розвитку, що супроводжується гіперурикемією. Також, можливе утворення каменів у нирках (нефролітіаз) або наявність крові у сечі (гематурія), викликані кислотно-сечовим камінням. Найчастіше підозри на синдром Леша-Ніхана виникають коли хворі причиняють самі собі поранення. Однак самотравмуюча поведінка зустрічається і в інших патологічних станах, таких як неспецифічна розумова відсталість, аутизм, синдром Туретта, синдром Корнелі де Ланж, синдром Айлі-Дея, нейрокантоцітоз, спадкові нейропатії першого типу, і деякі психіатричні захворювання. З перерахованого, тільки хворі з синдромом Леша-Ніхена, синдромом де Ланж, і синдромом Айлі-Дея, демонструють втрату тканин як наслідок самопоранень. Особливістю синдрому Леша-Ніхана, що відрізняє його від інших синдромів пов'язаних з нанесенням собі поранень, є кусання пальців, губ, внутрішньої поверхні щік. Наявність синдрому Леша-Ніхана повинно розглядатися тільки при наявності самотравмуючої поведінки разом з гіперурикемією і неврологічної дисфункцією.
Лікування
При синдромі Леша-Найхана лікування алопуринолом супроводжується зниженням рівня сечової кислоти (і зменшенням проявів подагричного артриту і відкладень солей); він неефективний відносно неврологічної симптоматики. У осіб з гіперурикозурією, що розвинулася внаслідок посилення синтезу сечової кислоти de novo або проведеної лікарської терапії, необхідно підтримувати досить високий об'єм сечі з рН 70.
Цього зазвичай досягають, використовуючи збалансовані суміші солей, такі як Polycitra, які більш ефективні, ніж гідрокарбонат. Важливість підтримки рН сечі на рівні близько 70 підтверджується тим фактом, що при рН 50 розчинність сечової кислоти становить 150 мг/л, в той час як при рН 70 - 2000 мг /л. Гіперурікемію при глікогенозі і, як і інші види вираженою гіперурикемії, необхідно коригувати, вона не піддається корекції при введенні пробенециду, але досить чутлива до впливу алопуринолу.
Синдром Дабіна — Джонсона
Синдром Дабіна-Джонсона — синдром спадкового ураження печінки, що проявляється розвитком жовтяниці, яка пов'язана з порушенням транспорту прямого (кон'югованого) білірубіну з гепатоцитів у жовч (екскреційна жовтяниця — варіант паренхіматозної жовтяниці). Синдром названий на честь американських патологоанатомів Ізідора Натана Дабіна та Франка Б. Джонсона.
Жінки хворіють частіше за чоловіків. Тривалість життя не відрізняється від здорових осіб, спеціального лікування не потребує. Прийом естрогенів при цьому захворюванні строго протипоказаний.
Етіологія
|
Р
|

Захворювання успадковується аутосомно-рецесивно (рис.12). Внаслідок мутації порушується повноцінне функціювання білка MRP2 (англ. multidrug resistance related protein 2), який відповідає за виділення глюкорунованого (кон'югованого) білірубіну в жовчні канальці. Внаслідок цього жовч не може повноцінно переходити з гепатоцитів в жовчні капіляри, що призводить до накопичення білірубіну у печінці і зворотного переходу кон'югованого (прямого) білірубіну до крові.
Клінічні ознаки
Синдром Дабіна-Джонсона проявляється жовтяницею з легкою інтермітуючою (епізодичною) гіпербілірубінемією. Підвищується рівень за рахунок прямого білірубіну (близько 60 %). У сечі присутній копропорфірин.
Діагностика
Поряд з лабораторними даними (білірубін, копропорфірин) вказує на хворобу коричнево-чорне забарвлення печінки «шоколадна печінка», яке виявляється при лапароскопії. Негативна холангіограма через порушення виділення рентгенконтрастної речовини.
Синдром Кріглера — Найяра
Синдром Кріглера—Найяра — сімейна форма вродженої гіпербілірубінемії, при якій відбуваються окрім жовтяниці тяжкі порушення центральної нервової системи (ЦНС), дегенерація базальних гангліїв. Це дуже рідке захворювання — її частота в світі складає 1:1 000 000.
Патогенез
Синдром спричинює дефект УДФ-глюкуронілтрансферази, що відповідає за кон'югацію білірубіну у печінковій клітині — гепатоциті. Внаслідок дефіциту цього ферменту непрямий білірубін не кон'югується у прямий. Розрізняють два типи хвороби:
Синдром Криглера—Найяра І типу (КГ-І) успадковується аутосомно-рецесивно і характеризується повною відсутністю ферменту глюкуронілтрансферази. Проявляється вираженою жовтяницею з явним переважання рівня прямого білірубіну у крові безпосередньо після народження. Діти з таким типом синдрому швидко гинуть.
Синдром Криглера—Найяра ІІ типу (КГ-ІІ, синдром Аріаса) — успадковується аутосомно-домінантно і відрізняється від першого типу тим, що внаслідок «пом'якшеної мутації» спостерігають часткове зниження активності ферменту, так що білірубін значною мірою кон'югується і виводиться з організму. Жовтяниця проявляється на першому році життя або навіть в підлітковому віці. Прогноз при цьому типі для хворих відносно сприятливий.
Патогенез
Тип І
При синдромі Криглера—Найяра типу І активність ферменту відсутня взагалі або наявні лише її сліди. Мутація розміщена в екзоні 2-5 гену УДФ-глюкорунілтрансферази, спричиняє утворення неповноцінного ферменту, який швидко руйнується. Внаслідок цього порушується процесс глюкуронування білірубіну, стероїдних гормонів і медикаментів. Тому концентрація некон'югованого білірубіну значно підвищується. Активність інших печінкових ферментів (амінотрансферази, γ-глутамілтранспептидази, лужної фосфатази) достатня, гістологія без особливостей. Жовч хворих майже безбарвна. При цьому типі синдрому не вдається досягти активації ферменту при прийомі ензимоіндукторів (фенобарбітал, рифампіцин, рифаксимін). Накопичений білірубін метаболізується дуже повільно і елімінується з калом у занадто малих об'ємах. У сечі уробіліноген не визначають.
Тип II
На противагу до першого типу при типі ІІ залишкова активність УДФ-глюкуронілтрансферази становить 10-20% від норми. Відповідальний за цей дефект гену уражає також УДФ-ГТ1-ген (існує понад 10 відомих форм мутації). Деякі форми можуть призводити до подібних І типом порушень кон'югації гормонів і медикаментів. Рівень некон'югованого білірубіну в крові підвищується при ІІ типі не так сильно. За допомогою фенобарбіталу можна підвищити активність ферменту і досягти рівня білірубіну в крові до 50 мкмоль/л/год. Як і при І типі розміри печінки і гістологія без особливостей.
Клінічні ознаки
Синдром Криглера—Найяра типу І проявляється безпосередньо після народження вираженою гіпербілрубунемією, яка при відсутності регулярного лікування призводить до розвитку ядерної жовтяниці з обтяжуючими неврологічними пошкодженнями.
Синдром Криглера—Найяра типу ІІ перебігає не так агресивно. Ядерна жовтяниця є рідким явищем. В подальшому житті жовтяниця і виражений свербіж шкіри можуть відчутно погіршити якість життя.
Лікування
Проводять з врахуванням типу і ступеня виразності хвороби. При типі І дуже важливим є ранній початок лікування. Етіологічне лікування полягає у вирішенні питання про трансплантацію печінки. При ІІ типі фенобарбітал, як єдиний на сьогодні ефективний печінковий індуктор ферментів для щоденного прийому, може допомогти знизити рівень білірубіну.
І тип
Консервативна терапія синдрому Криглера—Найяра типу І базується на трьох ступенях:
постійна щоденна фототерапія променями блакитного спектру;
прийом тінпротопорфірину, препарат знижує активність гемоксигенази;
прийом карбонату кальцію і фосфату кальцію (підвищує секрецію некон'югованого білірубіну у кишечнику).
При такому лікуванні можна подовжити тривалість життя і сповільнити розвиток неврологічних ускладнень, що дасть можливість провести трансплантацію печінки якомога раніше. В експериментальній фазі знаходиться алогенна трансплантація клітин печінки.
ІІ тип
Лікування типу ІІ досягається через щоденний прийом невеликих доз фенобарбіталу. Шляхом посилення ферментативної активності ферменту УДФ-глюкуронілтрансферази рівень білірубіну в крові може знижуватися до того рівня, що дає можливість людині жити досить повноцінним життям.
Хвороба Вільсона — Коновалова
Хвороба Вільсона (гепато-лентикулярна дегенерація, гепатоцеребральна дистрофія, псевдосклероз Вестфаля) — спадкове захворювання з аутосомно-рецесивним типом успадкування, при якому мідь накопичується у тканинах. Проявляється у вигляді неврологічних та/або психіатричних симптомів й уражень печінки. Хворобу Вільсона лікують медикаментами, які зменшують абсорбцію міді або видаляють її надлишок з організму. У деяких випадках необхідна трансплантація печінки.
Захворювання виникає через мутації у гені протеїну ATP7B, АТФ-ази P-типу, що транспортує катіони міді. Одна ненормальна копія цього гену наявна в 1 із 100 людей, у яких не розвинулись симптоми хвороби, — так званих носіїв хвороби. У дитини хвороба Вільсона може розвинутись лише у тому випадку, коли вона успадкує ген хвороби від обох батьків. Зазвичай симптоми починають проявлятися у віці від 6 до 20 років, але описані випадки у набагато старших людей. Хворобу Вільсона виявляють в 1-4 із 100 000 людей. Її названо на честь Самуеля Александра Кіннієра Вільсона.
Спадковість
Ген хвороби Вільсона (ATP7B) знаходиться у 13 хромосомі (ділянка 13q14.3) і його експресія в основному проявляється у печінці, нирках та плаценті. Ген кодує ATФазу P-типу (ензим, що транспортує катіони), що транспортує мідь у жовчі включає її до складу церулоплазміну. Мутації цього гену можна визначити у 90% випадках хвороби. Більшість з них (60%) є гомозиготами за мутацією ATP7B (дві аномальні копії), і 30% мають лише одну аномальну копію. У 10% хворих не відмічають мутацій.
Хоча описано лише 300 випадків мутації ATP7B гену, випадки хвороби Вільсона залежать від кількості мутацій, притаманних для певної популяції. Наприклад, у західних популяціях мутація H1069Q (заміщення гістидину глутаміном у білковій позиції 1069) присутня у 37—63% випадків, проте у Китаї ця мутація є дуже рідкісною, а мутація R778L (аргінін на лейцин у 778 позиції) трапляється набагато частіше. Достатньо мало відомо про вплив різних мутацій, хоча, згідно з деякими дослідженнями, мутації у H1069Q зазвичай відтерміновують початок симптомів.
Нормальні варіації у гені PRNP можуть модифікувати перебіг хвороби шляхом відтермінування віку початку симптомів та їх тип. Цей ген продукує PRNP (PRioN Protein), який проявляє активність у мозку та інших органах, а також бере участь у транспортуванні міді. До розвитку захворювання також може бути причетний ген ApoE, але дослідження ще не підтвердили цю теорію.
Захворювання успадковують за аутосомно-рецесивним типом. Для того щоб успадкувати його, обоє батьків мають бути носіями пошкодженого гену. Більшість хворих не вказують на родинну історію цією хвороби. Людей з лише однією анормальною копією гену називають носіями (гетерозиготи), і у них спостерігають незначні з медичної точки зору порушення метаболізму міді.
Хвороба Вільсона є найпоширенішою з групи спадкових захворювань, які спричиняють накопичення міді у печінці. Усі ці захворювання можуть спричинити цироз ще у ранньому віці. До цієї групи хвороб також відносять індійський дитячий цироз (ІДЦ), тирольський ендемічний інфантильний цироз та ідіопатичний мідний токсикоз. Ці хвороби не пов'язані з мутаціями у ATP7B, наприклад, ІДЦ пов'язують із мутаціями у генах KRT8 та KRT18.
Патогенез
Мідь виконує багато функцій в організмі. В основному вона виступає як кофактор для деяких ферментів, таких як церулоплазмін, цитохром C-оксидаза, дофамін-β-гідроксилаза, супероксиддисмутаза і тирозиназа.
Мідь всмоктується з шлунково-кишкового тракту. Транспортний білок на клітинах тонкої кишки CMT1 (Copper Membrane Transporter 1)переміщує мідь усередину клітин. Частина міді зв'язується з металотіонеїном, а інша — переміщується до комплексу Ґольджі за допомогою транспортного білка ATOX1. В апараті Ґольджі у відповідь на підвищення концентрації міді фермент ATP7A (Copper-transporting ATPase 1) вивільняє цей елемент через ворітну вену у печінку. У гепатоцитах білок ATP7B зв'язує мідь з церулоплазміном і вивільняє його у кров, а також видаляє надлишок міді з жовчю. Обидві функції ATP7B порушуються при хворобі Вільсона. Мідь накопичується у тканині печінки, церулоплазмін продовжує виділятись, але з нестачею міді (апоцерулоплазмін) швидко руйнується у кров'яному руслі.
Коли міді у печінці стає більше, ніж білків, які її зв'язують, відбувається їх окиснювальне пошкодження за рахунок реакції Фентона. Це призводить до запалення печінки, її фіброзу, і, як наслідок - до цирозу. Також з печінки у кров'яне русло виділяється мідь, незв'язана з церулоплазміном. Ця вільна мідь осідає по всьому організму, особливо в нирках, очах і головному мозку.
Основну роль у патогенезі грає порушення обміну міді, її накопичення у нервовій (особливо уражаються базальні ядра), нирковій, печінковій тканинах і рогівці ока, а також токсичне пошкодження міддю даних органів. Порушення метаболізму міді виражається у порушенні синтезу і зниженні у крові концентрації церулоплазміну. Церулоплазмін бере участь у процесі виведення міді з організму. У печінці формується великовузловий або змішаний цироз. У нирках у першу чергу страждають проксимальні канальці. У головному мозку уражаються більшою мірою базальні ядра, зубчасте ядро мозочка та чорна речовина. Відкладення міді в десцеметовій мембрані ока призводить до формування кілець Кейзера — Флайшера.
Гепалентикулярна дегенерація починається у дитячому або молодому віці і характеризується хронічним прогресивним перебігом. У багатьох випадках появі симптомів ураження нервової системи передують вісцеральні розлади у вигляді порушення роботи печінки і шлунково-кишкових розладів (жовтяниця, біль у правому підребер'ї, диспептичні явища). Іноді розвивається виражений гепато-спленальний синдром.
З боку нервової системи на перший план виступають екстрапірамідальні симптоми у вигляді м'язової ригідності, гіперкінезів і розладів психіки. Пірамідні симптоми можливі, але зазвичай відсутні. Чутливість, як правило, не порушується.
Розпізнають 5 форм хвороби Вільсона:
Черевна форма — тяжке ураження печінки, що призводить до смерті ще до появи симптомів з боку нервової системи; хворіють діти. Її тривалість коливається від декількох місяців до 3—5 років.
Ригідно-аритмогіперкінетична, або рання форма відрізняється швидким перебігом; починається навіть у дитячому віці. У клінічній картині переважає м'язова ригідність, що призводить до контрактур, сповільнення рухів, хореоатетоїдних або торсійних силових рухів. Характерними є дизартрія, дисфагія, судомний сміх і плач, аффективні розлади і помірне зниження інтелекту. Захворювання триває 2—3 роки, закінчується летально.
Тремтливо-ригідна форма зустрічається частіше решти; починається в юнацькому віці, перебігає повільніше, іноді з ремісіями і раптовими погіршеннями, що супроводжуються субфебрильною температурою. Характеризується одночасним розвитком важкої ригідності й тремтіння; тремтіння є дуже ритмічним (2—8 тремтінь на секунду), різко посилюється при статичному напруженні м'язів, рухах чи хвилюванні, у стані спокою під час сну зникає. Іноді можна виявити атетоїдні хореоформні силові рухи; спостерігаються також дисфагія і дизартрія. Середня тривалість життя становить близько шести років.
Тремтлива форма починається у віці 20—30 років, перебігає досить повільно (10—15 років і довше); тремтіння різко переважає, ригідність з`являється лише наприкінці хвороби, а інколи спостерігають гіпертонію м'язів; відмічають амімію, повільну монотонну мову, тяжкі зміни психіки, часто спостерігають афективні спалахи; можливі епілептиформні припадки.
Екстрапірамідно-кіркова форма зустрічається рідше решти форм. Типові для хвороби Вільсона порушення надалі ускладнюються пірамідними парезами, епілептиформними припадками і тяжким слабоумством (спостерігають обширні розм'якшення у корі великих півкуль). Триває 6—8 років, закінчується летально.
Патологічна анатомія
У головному мозку у разі гепато-церебральної дистрофії розм'якшується сочевицеподібне ядро, особливо скорпула, з утворенням дрібних кіст. Уражені й інші утвори: хвостате ядро, глибокі шари кори, мозочок, а зокрема зубчасті ядра, підгорбкові ядра; в інших ділянках головного мозку зміни виражені менше.
Усі зміни поділяють на ангіотоксичні та цитотоксичні. Перші проявляються атонією судин, особливо дрібних, та зміною їхніх стінок. У результаті виникають стази, поширений периваскулярний набряк з аноксією нервової тканини та її загибеллю часто спостерігають крововиливи та їхні сліди у вигляді скупчень гемосидерину.
Цитотоксичний компонент полягає у поширених дистрофічних змінах макроглії нервових клітин, що часто закінчується їх загибеллю. Характерним є поява глії Альцгеймера, що утворюється зі звичайних астроцитів. Нерідко зустрічають змінені нервові клітини, дуже схожі на альцгеймерську глію; схожі клітини виявляють також в печінці і нирках. В основі цих клітинних змін лежить фактор однотипного порушення клітинного обміну, імовірно, обміну нуклеїнових кислот.
Чим пізніше починається захворювання, тим повільніше воно перебігає, тим більш дифузними є зміни у головному мозку і тим більше цитотоксичний компонент переважає над ангіотоксичним. Як наслідок атрофічного цирозу, печінка є зменшеною і горбкуватою; ділянки нормальної тканини чергуються з некротичними, дегенеративними ділянками і з «острівцями» регенерації; швидке утворення судин призводить до появи анастомозів між гілками ворітної і нижньої порожнистої вен.
Ознаки і симптоми
Основними місцями накопичення міді є печінка та мозок, а тому захворювання печінки і нейропсихіатричні симптоми є основними ознаками, відповідно до яких ставлять діагноз. Люди з проблемами із печінкою (зазвичай це діти та підлітки) звертаються за медичною допомогою частіше, ніж ті, у кого наявні неврологічні та психіатричні симптоми (зазвичай це люди у віці 20 років та старші). У декого ідентифікують хворобу лише тому, що їхнім родичам поставили цей діагноз. У багатьох пацієнтів діагностують захворювання, і виявляється, що у них були наявні симптоми, але правильний діагноз їм не могли поставити.
Ураження печінки
Ураження печінки може проявлятись у вигляді втомлюваності, схильності до кровотеч та психічних порушень (через печінкову енцефалопатію) та портальної гіпертензії. Остання — це стан, при якому тиск у ворітній вені є значно підвищеним, що призводить до варикозів стравохідних та інших вен, збільшення селезінки (спленомегалії) та накопичення рідини у порожнині живота (асциту). При огляді такого пацієнта спостерігають такі ознаки ураження печінки, як павукоподібні ангіоми (малі роздуті патологічні анастомози кровоносних судин, зазвичай у навколоключичній ділянці). Хронічний гепатит з високим ступенем прогресування спричинює цироз печінки ще до появи симптомів. Хоча більшість хворих на цироз є у групі підвищеного ризику гепатоцелюлярної карциноми (пухлини печінки), у хворих на хворобу Вільсона цей ризик є відносно низьким.
Близько 5% усіх людей ставлять цей діагноз після виникнення у них гострої печінкової недостатності, часто у контексті гемолітичної анемії (анемія, яка виникає внаслідок руйнування еритроцитів). Це призводить до проблем в утворенні протеїнів (проявляється підвищеною коагуляцією) та порушень метаболізму у печінці. Пришвидшений метаболізм протеїнів призводить до накопичення продуктів обміну, таких як аміак, у кровоносному руслі. Коли це призводить до пошкодження мозку, то у людини розвивається хронічна печінкова енцефалопатія (психічні розлади, кома, судоми і життєво небезпечний набряк мозку).
Нейропсихіатричні ознаки
Приблизно половина людей з хворобою Вільсона мають неврологічні та психіатричні прояви. У більшості відзначають як легкі когнітивні порушення, так і зміни в поведінці. Зазвичай з часом виявляють нові симптоми, часто у вигляді паркінсонізму (брадикінезія та порушення балансу при ходьбі є звичайними ознаками паркінсонізму) та тремору рук, порушення експресії на обличчі, невиразної мови, атаксії (порушення координації) чи дистонії (скручувань та повторюваних рухів певної частини тіла). У деяких хворих спостерігають судоми та мігрень. У багатьох людей присутній так званий «wing-beating»-тремор; у решти людей він відсутній, проте його можна спровокувати, попросивши хворого витягнути руки вперед.
Хвороба Вільсона також може вплинути на когнітивні функції. Когнітивні розлади можуть мати двоякий генез: розлад лобової частки (може проявлятись у вигляді імпульсивності, апатії, порушенням можливості планувати та ставити цілі) та субкортикальна деменція (може проявлятись у вигляді сповільнення мислення, втрати пам'яті, без ознак афазії, апраксії чи агнозії). Є теорія, що ці когнітивні порушення напряму пов'язані з психічними розладами, які спричинює хвороба.
До психіатричних проблем, які зумовлює хвороба Вільсона, можна віднести зміни у поведінці, депресію, тривогу та психоз. Психіатричні зміни зазвичай проявляються разом із неврологічними симптомами і лише у рідкісних випадках без них. Часто ці симптоми важко визначити, або ж їм приписують інше походження. Саме через це рідко ставлять діагноз хвороби Вільсона за присутності лише психіатричних ознак.
Інші системи органів
Такі симптоми пов'язані з акумуляцією міді при хворобі Вільсона:
|
Р |
 ис.
13 – Кільця Кейзера
ис.
13 – Кільця КейзераНирки: нирковий тубулярний ацидоз (другого типу), дефект реабсобції бікарбонатів у проксимальних ниркових канальцях, що призводить до нефрокальцинозу (накопичення кальцію в нирках), ослаблення кісток (через втрату кальцію та фосфатів) і часом до аміноацидурії (екскреція незамінних амінокислот, потрібних для синтезу білків, з сечею).
Серце: кардіоміопатія є рідкісним симптомом хвороби Вільсона; вона може призвести до серцевої недостатності (порушення насосної функції) та аритмій.
Гормони: гіпопаратироїдизм, безпліддя та викидні.
Діагностика
Хворобу Вільсона можна запідозрити, ґрунтуючись на будь-якому з вище перерахованих симптомів або ж у випадку, коли у близького родича діагностовано це захворювання. У більшості пацієнтів спостерігають легку форму дисфункції печінки, яку при біохімічному дослідженні підтверджує підвищений рівень білірубіну в крові, висока активність аспартатамінотрасферази та аланінамінотрансферази. При тяжких ушкодженнях печінки можна спостерігати знижений рівень альбуміну через неспроможність ушкоджених гепатоцитів синтезувати цей клас білків; також протромбіновий час може бути подовженим, оскільки печінка втрачає здатність продукувати фактори зсідання крові. Активність ферменту лужної фосфатази є відносно низькою у хворих із печінковою недостатністю, яку спричинює хвороба Вільсона. Пацієнтам з неврологічними симптомами зазвичай проводять магнітно-резонансну томографію, що дає змогу побачити підвищену активність базальних ядер за часом спінової релаксації. Також МРТ може показати характерний візерунок «обличчя великої панди».
Немає прямого тесту для діагностування хвороби Вільсона, проте рівень церулоплазміну та міді у крові, а також кількість міді, що виводиться з сечею за 24 години, разом допомагають скласти висновок про кількість міді в організмі. «Золотим стандартом» чи найкращим аналізом є біопсія печінки.
Рівень церулоплазміну є анормально низьким (<0,2 г/л) у 80—95% випадків. Проте він може бути у межах норми в людей із запальними процесами в організмі, оскільки церулоплазмін є «стрес-білком». Низький рівень церулоплазміну також притаманний хворобі Менкеса та ацерулоплазмінемії, які є пов'язаними з цим явищем, проте ця ознака характерніша для хвороби Вільсона.
Поєднання неврологічних симптомів, кілець Кейзера — Флайшера та низький рівень церулоплазміну є чільними ознаками, що вказують на хворобу Вільсона. У багатьох випадках для уточнення діагнозу потрібні додаткові аналізи.
Мідь у крові та сечі
Рівень міді у крові є низьким, що може видатись парадоксальним, оскільки хвороба Вільсона — це захворювання надлишку міді. Однак 95% міді у плазмі крові переносить церулоплазмін, рівень якого є звичайно низьким при хворобі Вільсона. Для діагностики також проводять забір сечі, яку зберігають у посудині, що не містить у своєму складі міді. За 24 години перевіряють рівень міді: вище 100 мкг/24год (1,6 мкмоль/24год) підтверджує діагноз хвороби, а рівень вище 40 мкг/24год (0,6 мкмоль/24год) є зоною ризику виникнення цієї хвороби. Високий рівень міді у сечі не є унікальною (патогномонічною) ознакою хвороби Вільсона, натомість це явище також спостерігають при автоімунному гепатиті та холестазі (а також будь-якій іншій хворобі, що перешкоджає потраплянню жовчі у дванадцятипалу кишку.
Для діагностики у дітей інколи використовують пеніциламіновий аналіз. Дитина споживає 500 мг пеніциламіну перорально, і після цього проводять забір сечі протягом 24 годин. Рівень вище 1600 мкг (25 мкмоль) свідчить на користь хвороби Вільсона. Дорослим такий тест не проводять.
Біопсія печінки
Гістохімічні методи визначення рівня міді у крові чи сечі самі по собі є ненадійними і завжди потребують проведення додаткових аналізів. Тож після підтвердження хвороби Вільсона усіма доступними тестами, вирішальним дослідженням є забір маленької частинки печінкової тканини шляхом біопсії печінки. Печінкову тканину досліджують патоморфологічно — мікроскопічним шляхом на перевірку рівня стеатозу, цирозу та кількості акумульованої міді. Рівень 250 мкг міді на грам зневодненої тканини печінки підтверджує хворобу Вільсона. Зрідка спостерігають низький рівень міді; у такому випадку сукупність даних біопсії та інших досліджень все ж дає змогу поставити діагноз гепатоцеребральної дистрофії.
На ранніх стадіях захворювання біопсія звичайно знаходить значний стеатоз (накопичення ліпідів у клітині), підвищений вміст глікогену в гепатоцитах та ділянки некрозу (смерті клітин). У пізніших стадіях захворювання, спостерігають зміни, схожі зі змінами при автоімунному гепатиті: інфільтрація запаленими клітинами, частковий некроз та фіброз. На останніх стадіях захворювання, як правило, біопсія виявляє цироз.
Генетичний аналіз
Аналіз мутації генуATP7B та решти генів, пов'язаних з накопиченням міді в печінці, є варіативним при діагностиці захворювання. У випадку підтвердження мутації варто перевірити близьких родичів хворого на присутність прихованого захворювання. Важливим фактором при аналізі геному є різниця у поширенні генів хвороби Вільсона за регіонами планети.
Лікування
Дієта
Загалом, рекомендують харчуватись їжею з низьким вмістом міді, уникати споживання грибів, горіхів, шоколаду, сухофруктів, печінки та молюсків.
Медикаментозна терапія має бути спрямована за двома основними механізмами: одні підвищують екскрецію міді з організму, а інші натомість зменшують всмоктування міді з продуктів харчування у кишці.
Основою лікування є призначення пеніциламіну. Він зв'язує мідь, що призводить до виведення її з сечею. Таким чином, моніторинг кількості міді у сечі може бути корисним для перевірки ефективності лікувального процесу. Проте у 20 % випадків вживання пеніциламіну відзначають побічні ефекти, такі, як медикаментозний вовчак (що спричинює біль у суглобах та висип на шкірі) чи міастенію (нервовий розлад, що породжує м'язову слабкість). У хворих з неврологічною формою захворювання у половині випадків спостерігають парадоксальне погіршення симптомів. Хоча цей феномен характерний і для інших засобів лікування хвороби Вільсона, у цьому випадку зупиняють прийом пеніциламіну і призначають інші препарати. Хворим зі стійкістю до пеніциламіну часто призначають триентину гідрохлорид, який також має хелативні властивості. Багато лікарів рекомендують триентин як основний препарат, проте використання пеніциламіну все ж є ширшим. До інших ліків, що зв'язують мідь у кишечнику є тетраетилмолібдат, проте його використання є ще на етапі експериментального дослідження, але деякі дослідники вже повідомляють про його позитивний ефект.
Після повернення усіх показників до норми, хворим призначають цинк (зазвичай у вигляді препарату ацетату цинку) на заміну хелаторам для забезпечення сталого рівня міді в організмі. Цинк стимулює металотіонеїн, білок ентероцитів, що зв'язує мідь та запобігає її всмоктуванню і, відповідно, транспорту до печінки. Цинкову терапію проводять допоки не рецидивують симптоми хвороби або рівень міді у сечі не підвищиться.
У рідкісних випадках тяжкої неврологічної форми захворювання, коли ліки для перорального застосування не є ефективними, використовують препарат димеркаптол. Його вводять внутрішньом'язово декілька тижнів. Димеркаптол має низку неприємних побічних ефектів, зокрема, таких як біль у місці ін'єкції.
Людям з латентною формою хвороби проводять загальний курс лікування, оскільки накопичення міді може принести шкоду організму у довгостроковій перспективі.
Фізіотерапія
Фізіотерапія є корисною для пацієнтів з неврологічною формою захворювання. Лікування хелаторами міді може тривати до шести місяців до початку отримання ефекту, а фізіотерапія може допомогти боротись з атаксією, дистонією та тремором, а також запобігти розвитку контрактур, що можуть перейти у дистонію.
Трансплантація
Трансплантація печінки є ефективним засобом для лікування хвороби Вільсона, проте використовують її лише в окремих випадках, що пов'язано з ризиком даної процедури. Як правило, її проводять людям із гострою печінковою недостатністю, що не піддається медикаментозному лікуванню або людям із розвиненою хронічною печінковою недостатністю. Трансплантацію печінки не проводять пацієнтам із тяжкими нейропсихіатричними захворюваннями, при яких ця операція не дасть вагомої користі здоров'ю.
Прогноз
За відсутності ефективного лікування хвороба Вільсона швидко прогресує і зрештою призводить до смерті. Якщо вдається вчасно діагностувати захворювання та розпочати лікування, то у більшості хворих не змінюється їх якість життя і вони мають змогу продовжувати жити повною мірою.
