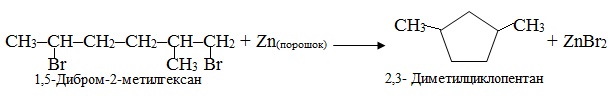- 6.1 Особливості реакцій між органічними сполуками
- 6.2 Класифікація органічних реакцій за структурними ознаками
- 6.3 Класифікація за валентним станом атомів Карбону
- 6.4 Класифікація за природою реагентів
- 6.5 Приклади розв'язання типових задач
Ключові терміни:
cхема реакції, α-елімінування, β-елімінування, γ-елімінування, π-комплекс, σ-комплекс, алкілювання, аренонієвий катіон, ароматизація, ацетилювання, ацилювання, вихід реакції, вільний радикал, віцінальні дигалогенопохідні, галогенування, гемінальні дигалогеновуглеводні, гідратація, гідрогалогенування, гідрування, дегалогенування, дегідратація, дегідрогалогенування, дегідрування, декарбоксилювання, десульфування, електрофільне заміщення, електрофільне приєднання, елементарна ланка, елементарна стадія, еноли, карбкатіон, карбоксилювання, карбонілювання, кето-енольна таутомерія, ланцюгові реакції, лужний гідроліз, механізм реакції, мономер, нуклеофільне заміщення, нуклеофільне приєднання, нітрування, оксонієвий аніон, оксонієвий катіон, олігомер, олігомерізація, пероксидний ефект Хараша, пероксидний ефект Харраша, поліконденсація, полімер, полімеризація, правило Ельтекова, правило Зайцева, правило Марковникова, правило Хюккеля, радикальне заміщення, радикальне приєднання, реагент, реакція Вюрця, реакція Дюма, реакція Зелінського-Казанського, реакція Кольбе, реакція Фріделя-Крафтса, реакція естерифікації, реакція лужного плавлення, реакції перегрупування, сополімеризація, ступінь полімеризації, субстрат, сульфування, суха перегонка солей, формілювання, ізомеризація6.1 Особливості реакцій між органічними сполуками
Реакції між органічними сполуками підлягають практично тим же самим законам, що і реакції неорганічних речовин, але в той же час мають деякі свої специфічні особливості.
- В неорганічних реакціях, особливо тих, перебіг яких проходить у розчинах чи розплавах, найчастіше беруть участь іони, а при взаємодії органічних сполук в реакцію вступають переважно молекули; при цьому відбувається розрив старих ковалентних зв’язків і утворення нових.
- Неорганічні реакції звичайно перебігають швидко, інколи миттєво, а взаємодія між органічними сполуками відбувається повільно, іноді десятки годин; для їх успішного завершення майже завжди необхідно застосовувати жорсткі умови: підвищені температури, тиск, випромінювання, каталізатори.
- Неорганічні реакції проходять до повного витрачання вихідних реагентів (необоротні процеси) чи до досягнення стану рівноваги (оборотні процеси), а органічні реакції рідко закінчуються високим виходом продуктів: у більшості випадків він не перебільшує 40-50%.
Вихід реакції η – це частка реально одержаного продукту від теоретично розрахованого; виражається в частках одиниці чи у відсотках:
![]()
- В органічних реакціях спостерігається, як правило, паралельний перебіг декількох процесів, внаслідок чого одержують суміш ізомерів чи певну кількість побічних продуктів. З цієї причини не завжди доцільно користуватися рівняннями реакцій, які виражають стехіометричні кількісні співвідношення між вихідними речовинами і продуктами. В органічній хімії частіше звертаються до схем.
Схема реакції – це умовний запис взаємодії між сполуками, в якому без урахування коефіцієнтів зазначають вихідні речовини і основні продукти, а над стрілкою – умови перебігу реакції (температура, тиск, каталізатор). Наприклад, схема нітрування толуену не показує кількісного співвідношення між вихідними і кінцевими речовинами:
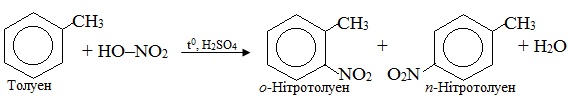
Цю ж схему реакції можна записати і так:
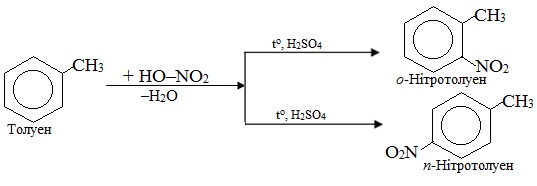
Необхідно уточнити, що в органічній хімії поряд з терміном «вихідні речовини» розповсюджені й інші: субстрат і реагент.
- Cубстрат – це речовина, реакційна здатність якої вивчається в даній реакції або яка є основою для одержання головного продукту реакції. Реагент – сполука, якою діють на субстрат для дослідження його властивостей.
Так, в розглянутій вище реакції роль субстрату виконує толуен, а реагенту – нітратна кислота. Проте при взаємодії рівнозначних вихідних речовин поняття субстрату і реагенту втрачають зміст.
6.2 Класифікація органічних реакцій за структурними ознаками
Величезна кількість органічних реакцій, які характеризуються різними ознаками, сприяє існуванню достатньо великої кількості класифікацій. При цьому, залежно від ознаки, яку беруть до уваги, одна і та сама органічна реакція може одночасно належати до декількох типів. Більшість процесів піддається класифікації, що значно полегшує їх вивчення, однак в органічній хімії зустрічаються складні реакції, які неможливо віднести до якоїсь конкретної групи.
Важливішою структурною ознакою органічних реакцій є природа (тобто – склад) реагенту, залежно від чого розглядають певні типи органічних реакцій: приєднання, заміщення, елімінування, перегрупування, полімеризації. Однак у свою чергу кожний тип реакцій може проходити залежно від природи атакуючої частинки за різними механізмами – електрофільним, нуклеофільним чи радикальним.
Механізм реакції – це сукупність і послідовність елементарних стадій, через які проходить хімічна реакція від вихідних речовин до кінцевих продуктів. Елементарна стадія – це проміжний одиничний процес протягом хімічної реакції, який не може бути розділеним на простіші акти хімічної взаємодії.
6.2.1 Реакції приєднання
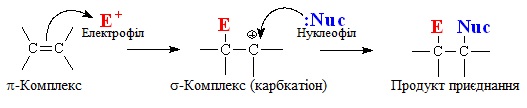
Реакціями приєднання називають такі хімічні процеси, при яких відбувається сполучення субстрату, в молекулі якого містяться кратні зв’язки, і реагенту. Реакції приєднання позначають символом А (від англ. addition – додавання), вони поділяються на певні групи залежно від механізму перебігу.
1. Електрофільне приєднання АЕ, для позначення якого поруч з символом А дописується індекс Е, що вказує на електрофільну природу реагенту (див.§5.3 ). Субстратом в реакціях АЕ найчастіше бувають сполуки, що містять кратні зв’язки – подвійні чи потрійні. Наприклад, за механізмом електрофільного приєднання АЕ (рис. 6.1) відбувається реакція бромування етену, внаслідок якої утворюються віцінальні дигалогенопохідні (тобто такі, в яких атоми Hal сполучені з двома сусідніми атомами С):
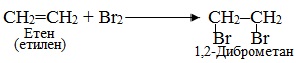
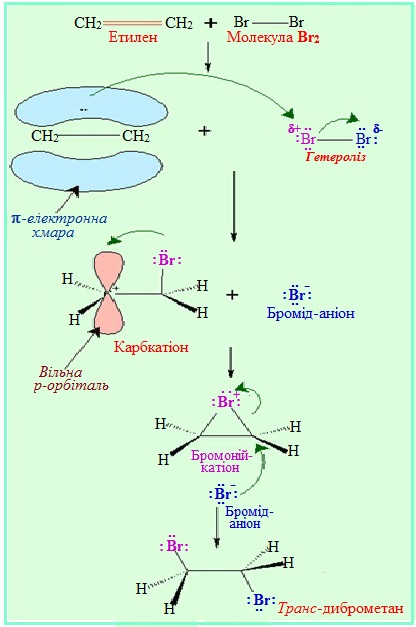
Рисунок 6.1 – Механізм реакції електрофільного приєднання АЕ при бромуванні етену (етилену)
У випадку несиметричної будови субстрату напрямок електрофільного приєднання до нього реагентів, що складаються з полярних молекул (H2O, HHal, HNH2), визначається відповідно до правила Марковникова.
Правило Марковникова: При взаємодії несиметричних ненасичених сполук з полярними молекулами типа НХ атом Гідрогену приєднується до найбільш гідрогенізованого атома Карбону (тобто сполученого із більшою кількістю атомів Н), який утворює кратний зв'язок.
Як приклад можна навести реакцію гідробромування 2-метилбут-2-ену (тобто приєднання до субстрату С5Н10 гідрогенброміду HBr):
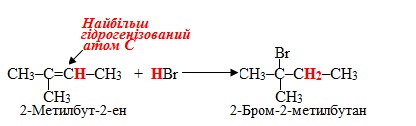
При електрофільному приєднанні електронна густина π-зв'язку стає об'єктом атаки електрофільних реагентів, що спричиняє гетеролітичний розрив зв'язку. Весь процес відбувається за декількома стадіями (рис. 6.2).
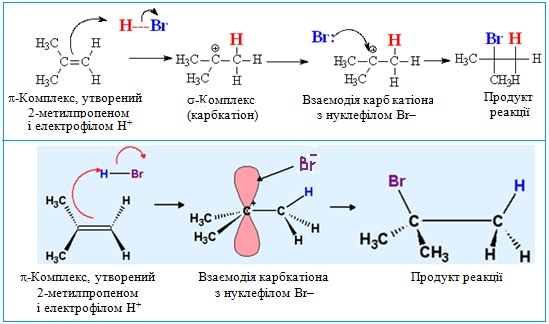
Рисунок 6.2 – Варіанти схематичного зображення механізму реакції електрофільного приєднання АЕ на прикладі гідробромування 2-метилпропену
Перша стадія – взаємодія між атакуючою електрофільною частинкою Е+ і електронною хмарою π-зв'язку. В результаті електростатичного притягання утворюється так званий π-комплекс – проміжна нестійка система. Достатньо часто електрофільною частинкою виступає протон (тобто катіон Гідрогену Н+, джерелом якого є кислоти HCl, H2SO4, HBr тощо). Графічно виникнення π-комплексу зображують за допомогою стрілки, напрямленої у бік електрофілу:
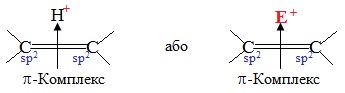
Друга стадія – виникнення σ-зв'язку між електрофілом Е+ (або Н+) і одним з атомів Карбону при подвійному зв’язку (>C=C<). Для утворення нового σ-зв’язку електрофіл «витягує» два електрони з π-зв'язку, що належали обом атомам С. Атом Карбону, що утворив зв’язок С–Н, переходить в стан sp3-гібридизації, а сполучений з ним атом С, який втратив електрон з рz-орбіталі, залишається в sp2-гібридизованому стані і набуває позитивного заряду (С+). Отже, утворюється карбкатіон (іон, в якому позитивний заряд зосереджений на атомі Карбону), який ще називають σ-комплекс:
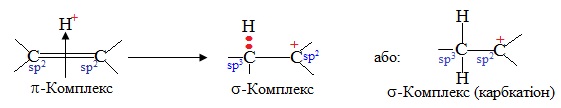
Третя стадія – взаємодія карбкатіона з нуклеофілом :Nuc (або будь-яким аніоном Х–, що міститься в реакційному середовищі), і утворення з ним нового σ-зв'язку за рахунок неподіленої електронної пари нуклеофілу (аніона).
У загальному вигляді механізм реакції електрофільного приєднання АЕ можна зобразити за допомогою схеми:
2. Нуклеофільне приєднання (АN), при якому реагуючою частинкою є нуклеофіл (:Nuc, або аніон Х–). Механізм реакції нуклеофільного приєднання АN розглянемо на прикладі карбонільних оксосполук – альдегідів і кетонів:

В оксосполуках полярність зв`язку С=О забезпечує виникнення часткових зарядів на атомах Карбону і Оксигену (Сσ+ і Оσ–) в карбонільній групі >С=О. Реакція за механізмом нуклеофільного приєднання АN за участю оксосполук залежно від умов проходить за однією із схем:
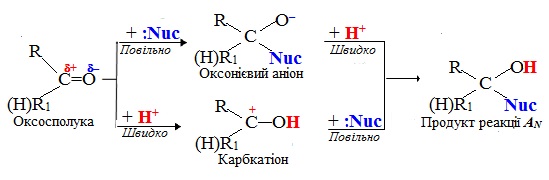
Якщо атакуючою частинкою є великий за розміром нуклеофіл (верхня гілка на схемі), він досить повільно приєднується до атома Сδ+ карбонільної групи, розриваючи подвійний зв’язок С=О. Внаслідок цього карбонільний атом С переходить у sp3-гібридизований стан і утворюється оксонієвий аніон з негативним зарядом на атомі О. На наступному етапі протон Н+ дуже швидко приєднується до атома О– і утворюється продукт реакції. Послідовність стадій змінюється в тому випадку, коли реакція каталізується мінеральними кислотами чи проходить у кислому середовищі (нижня гілка на схемі). Спочатку протон кислоти (Н+) дуже швидко приєднується до атома Оδ–, внаслідок чого на атомі Карбону виникає позитивний заряд (С+) – проміжна частинка стає карбкатіоном. На другій, повільній, стадії відбувається взаємодія карбкатіона з нуклеофілом (:Nu). Незважаючи на те, що процес формально починається з електрофільної стадії (атакуючою частинкою є Н+), реакція в цілому проходить за механізмом нуклеофільного приєднання АN, оскільки лімітуючою (найповільнішою) стадією, що визначає швидкість реакції в цілому, є саме приєднання нуклеофілу до карбкатіона.
3. Радикальне приєднання АR, при якому реагентом є вільний радикал – частинка з неспареним електроном. Механізм реакції радикального приєднання АR розглянемо на прикладі гідробромування несиметричного алкену за наявності пероксиду Н2О2:
![]()
В загальному випадку взаємодія ненасичених вуглеводнів з HBr та іншими гідрогенгалогенідами (HF, HCl, HI) проходить за протилежним шляхом – відповідно до правила Марковникова. Проте в присутності пероксидних сполук (Н2О2, С6Н5СОООН, Na2O2) діє пероксидний ефект Хараша: приєднання HBr до кратного зв’язку відбувається всупереч правилу Марковникова. Це означає, що атом Br сполучається з більш гідрогенізованим атомом С кратного зв’язку (тобто з атомом С, сполученим з більшою кількістю атомів Н).
Механізм складається з трьох стадій.
Перша стадія – ініціювання ланцюгу – може розпочинатися спонтанно чи примусово (шляхом фотохімічного, електрохімічного, термічного впливу або хімічним способом). При цьому пероксид розкладається на два вільні радикали:
![]()
Завдяки надзвичайно високій реакційній здатності радикал R–O• миттєво відщеплює атом Н від молекули реагенту HBr, утворюючи вільний радикал брому:
![]()
Скорочено сутність першої стадії відображає рівняння:
![]()
Друга стадія – розвиток ланцюгу, під час якого π-зв'язок в алкені розривається гомолітично, радикал Br• приєднується до більш гідрогенізованого атома С, а на менш гідрогенізованому атомі С залишається неспарений електрон. Таким чином утворюється вторинний вільний радикал. Утворення первинного несиметричного первинного радикалу, в якому неспарений електрон належить крайньому атому С, енергетично невигідно:
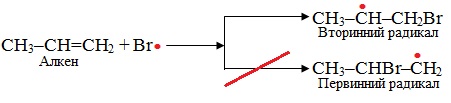
Вільний вторинний радикал взаємодіє з молекулою HBr, приєднуючи до себе атом Гідрогену з неспареним електроном і утворюючи радикал Br·, який знов вступає в реакцію з вихідним алкеном і т.д. – починається ланцюговий процес:
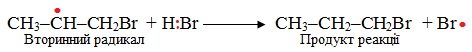
Третя стадія – обрив ланцюгу може трапитися у будь-який час при випадковому зіткненні двох радикалів, наприклад:
![]()
6.2.2 Реакції заміщення
В реакціях заміщення складова частина субстрату замінюється складовою частиною реагенту, наприклад:
Реакції заміщення позначають символом S (від англ. substitution – заміщення), вони поділяються на певні групи залежно від механізму перебігу.
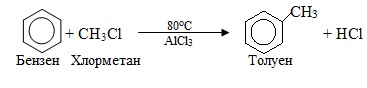
1. Електрофільне заміщення SE, при якому атакуючою частинкою є електрофіл. Найчастіше механізм SE реалізується в реакціях за участю ароматичних сполук, наприклад:
Ароматичними називають сполуки, які об'єднує сукупність певних ознак відповідно до правило Хюккеля: ароматичність – це наявність замкнутої спряженої системи, що має плоский циклічний σ-скелет і 4n+2 усуспільнені π-електрони, де n = 1, 2, 3…
Механізм електрофільного заміщення складається з декількох послідовних стадій.
Спочатку відбувається генерування електрофільної частинки у відповідних умовах під впливом каталізатора, який сприяє спочатку поляризації, а потім і розриву зв'язків у молекулі реагенту:
![]()
Далі дуже швидко, без порушення ароматичної системи, утворюється π-комплекс – проміжний стан, при якому спостерігається електростатичне притягання електрофілу Е+ до π-електронної хмари бензенового кільця:
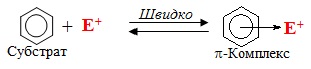
На наступній повільній стадії виникає σ-комплекс – карбкатіон, який у випадку ароматичних сполук називають ще аренонієвий катіон, що несе позитивний заряд, зосереджений на бензеновому кільці. В аренонієвому катіоні електрофіл Е+ сполучений σ-зв'язком з одним з атомів Карбону за рахунок двох π-електронів, що вилучаються із шестиелектронної спряженої системи. Цей атом Карбону переходить з sp2- у sp3-гібридизований стан, тому ароматичність системи порушується, оскільки в кільці залишається тільки чотири π-електрони, що розподілені на п'ять sp2-гібридизованих атомів С:
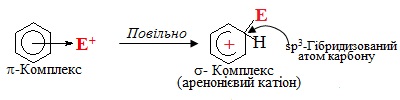
Подальший етап механізму електрофільного заміщення SE – відтворення ароматичності. Оскільки втрата ароматичності енергетично невигідна, система прагне повернути її найпростішим шляхом, а саме – відщепленням протону Н+ від σ-комплексу. Внаслідок цього два електрони, що утворювали σ-зв'язок С–Н, приєднуються до тих чотирьох π-електронів, які залишалися в σ-комплексі. Тому замкнута шестиелектронна система поновлюється і молекула переходить в ароматичний стан:
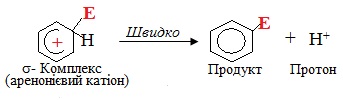
Одразу після цього утворюється побічний продукт НА: відщеплений протон Н+ взаємодіє з негативно зарядженим аніоном А–, що утворився при гетеролітичному розриві зв'язків у молекулі реагенту на перший стадії:
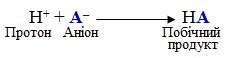
У загальному вигляді механізм реакції електрофільного заміщення SE за участю аренів зображується схемою:
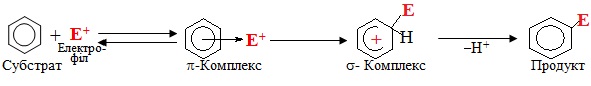
Як приклад реакції електрофільного заміщення SE можна навести бромування бензену:
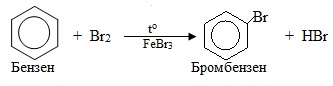
Спочатку під дією каталізатора FeBr3 відбувається гетеролітичний розрив зв’язку в молекулі реагенту (Br–Br) і утворення електрофільної частинки Br+ (рис. 6.3а). При наближенні до бензену електрофіл (Br+) дає проміжний π-комплекс, який існує за рахунок електростатичного притягання електрофільної частинки до делокалізованої π-електронної густини бензенового кільця. Поступово ароматичність порушується, при цьому один з атомів С сполучається σ-зв’язком з електрофілом Br+ (за рахунок вилучення двох π-електронів з шестиелектронної делокалізованної π-хмари) і одночасно переходить у sp2-гібридизований стан – виникає неароматичний σ-комплекс. А далі ароматичність відтворюється при відщепленні протона Н+ і утворенні продуктів реакції (рис. 6.3б).
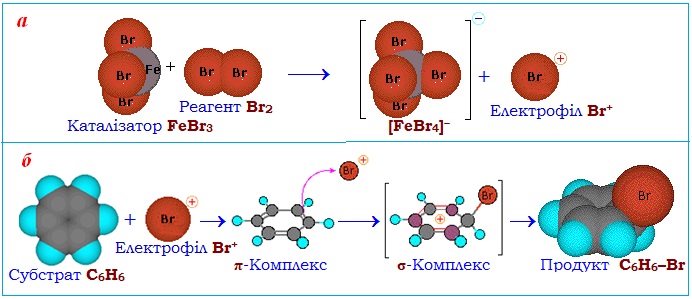
Рисунок 6.3 – Модель реакції бромування бензену за механізмом SE: а) утворення електрофілу Br+ при гетеролітичному розриві зв’язку Br–Br в молекулі реагенту Br2 під дією каталізатора FeBr3; б) взаємодія субстрату (бензену С6Н6) з електрофілом Br+, послідовне виникнення π-і σ-комплексів і утворення основного продукту реакції – бромбензену С6Н5–Br
2. Нуклеофільне заміщення SN, при якому атакуючою частинкою є нуклеофіл. Механізми реакціїй нуклеофільного заміщення можуть дещо різнитися, тому розглядають два основних типи.
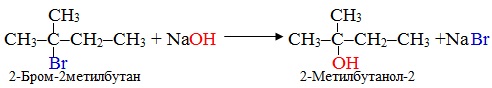
2.1. Механізм мономолекулярного нуклеофільного заміщення SN1. Термін «мономолекулярне заміщення SN1» означає, що швидкість реакції залежить від концентрації тільки одного реагенту. Механізм SN1 звичайно зустрічається в реакціях третинних галогеналканів (СnН2n+1)3С–Hal і третинних одноатомних спиртів (СnН2n+1)3С–ОН (тобто таких, в яких замісник сполучений з третинним атомом Карбону).
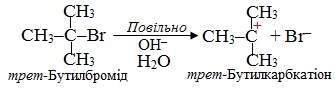
Механізм мономолекулярного нуклеофільного заміщення SN1 складається з декількох стадій, сутність яких розглянемо на прикладі реакції третинного галогеналкану з водним розчином лугу (або з водою в лужному середовищі) – так званий лужний гідроліз:
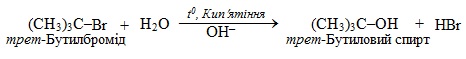
На першій, найповільнішій, стадії під дією полярного реагенту відбувається іонізація (тобто розклад на іони) молекули субстрату з утворенням карбкатіона:
На другій стадії, що проходить дуже швидко, карбкатіон взаємодіє з нуклеофілом – Н2О, в якому на атомі О є неподілені електронні пари. При цьому утворюється оксонієвий катіон – частинка, в якій позитивний заряд зосереджується на атомі Оксигену:
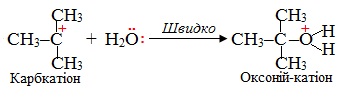
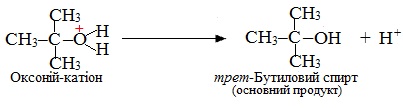
На останній стадії від оксонієвого катіона відщеплюється протон Н+ і утворюються кінцеві продукти реакції: основний продукт – спирт і побічний продукт – бромоводень у результаті сполучення протону Н+ з аніоном Br–:
За механізмом нуклеофільного мономолекулярного заміщення SN1 проходять і реакції за участю алілгалогенідів
CH2=CH–CH2–Hal і бензилгалогенідів C6H5–CH2–Hal:
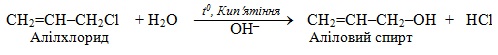
2.2. Механізм бімолекулярного нуклеофільного заміщення SN2, при якому швидкість реакції залежить від концентрації обох вихідних речовин. Реакція складається з однієї поступової стадії (рис. 6.4), під час якої в перехідному стані одразу проходять синхронно два процеси: розрив старого зв'язку С–Hal і утворення нового зв'язку C–Nuc між атомом Карбону і нуклеофілом:
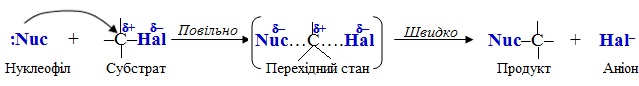
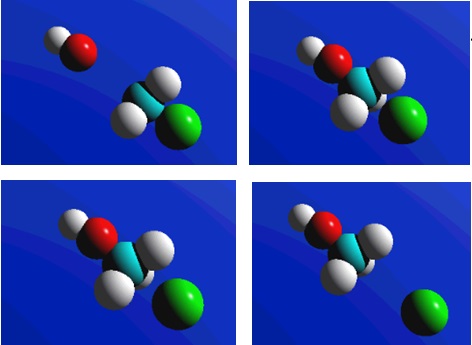
Рисунок 6.4 – Модель механізму реакції бімолекулярного нуклеофільного заміщення SN2: червоним кольором позначено нуклеофільну ділянку реагенту, блакитним – електрофільний центр молекули субстрату, зеленим – фрагмент молекули субстрату, заміщення якого відбувається
Механізм SN2 є характерним для первинних і вторинних галогеналканів при їх взаємодії з нуклеофільними реагентами. Важливе значення в органічному синтезі мають реакції лужного гідролізу полігалогенопохідних. Наприклад, гемінальні дигалогеновуглеводні (назва вказує, що обидва атоми галогену сполучені з одним атомом С) внаслідок лужного гідролізу дають карбонільні сполуки (альдегіди чи кетони):
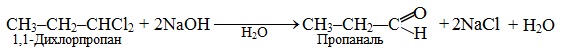
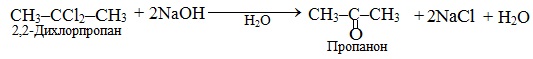
А тригалогенозаміщені гемінальні алкани при лужному гідролізі можуть перетворюватися на відповідні карбонові кислоти чи їх солі:
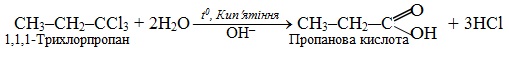

Віцінальні дигалогенопохідні, в яких два атоми галогену сполучені з двома сусідніми атомами С, та ізольовані дигалогенопохідні внаслідок лужного гідролізу приводять до двохатомних спиртів:
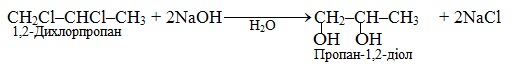
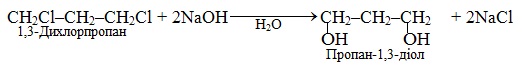
Цікаво поводять себе по відношенню до водних розчинів лугів вініл- і арилгалогеніди. Вінільні галогенопохідні швидко ізомеризуються у відповідні альдегіди згідно з правило Ельтекова (докладніше див. § 6.2.4):
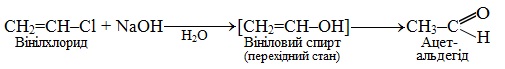
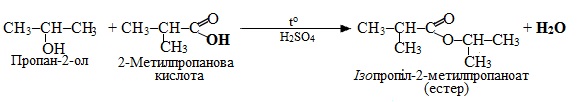
Механізм бімолекулярного нуклеофільного заміщення SN2 розглянемо на прикладі реакції естерифікації – у простішому випадку це взаємодія кислоти з спиртом за наявності каталітичної кількості сильної мінеральної кислоти (Н+), внаслідок чого утворюються естери (рис. 6.5) відповідно до загальної схеми:
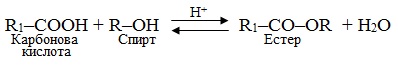
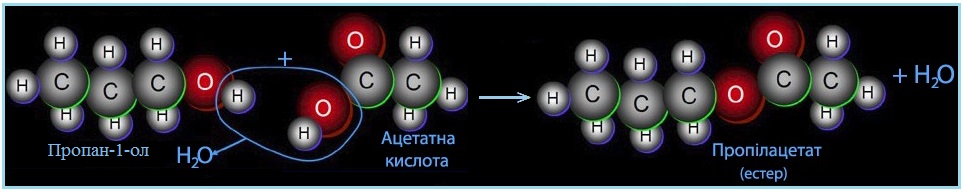
Рисунок 6.5 – Спрощена модель реакції естерифікації за механізмом бімолекулярного нуклеофільного заміщення SN2
Механізм реакції естерифікації за типом бімолекулярного нуклеофільного заміщення SN2 описується загальною схемою:
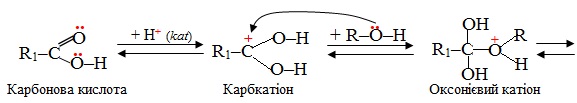
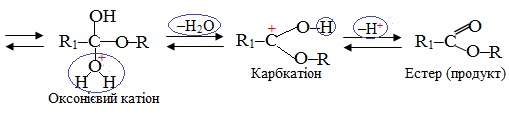
Реакція естерифікації може проходити і при взаємодії алканолів з мінеральними кислотами, наприклад:

З багатоосновними мінеральними кислотами утворюються неповні або повні естери, як показано на прикладі взаємодії етанолу з сульфатною кислотою (формулу H2SO4 у рівняннях органічних реакцій найчастіше записують у вигляді HO–SO3H чи навіть HO–SO2–ОH):
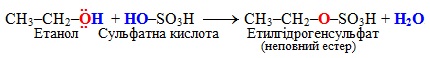
![]()
Слід запам’ятати, що необхідно розрізняти неповний естер і сульфокислоти:

3. Радикальне заміщення SR, при якому атакуючою частинкою є вільний радикал – частинка з підвищеною реакційною активністю, на валентній орбіталі якої міститься неспарений електрон. Вільні радикали завдяки високій реакційній здатності можуть існувати протягом дуже короткого часу (≈10–7 с). За механізмом радикального заміщення відбуваються ланцюгові реакції насичених вуглеводнів з галогенами при опромінюванні (hν) чи нагріванні. Механізм реакції радикального заміщення SR складається з трьох стадій, які зручно розглянути на прикладі хлорування метану:
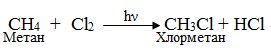
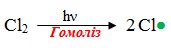
Перша стадія – ініціювання ланцюгу, під час якої завдяки опромінюванню відбувається гомоліз молекули галогену (розщеплення на два вільних радикали, в яких неспарений електрон зображується точкою):
Друга стадія – зростання ланцюгу. Радикал Cl• атакує молекулу алкана, вириває з неї атом Гідрогену і утворює нейтральну молекулу хлороводню і новий радикал (•СН3), виникнення якого забезпечується гомолітичним розривом зв'язку С–Н в метані:
![]()
У метильному радикалі (•СН3) атом Карбону переходить в стан sp2-гібридизації. Три sp2-гібридизовані орбіталі перекриваються з s-орбіталями трьох атомів Н, а на негібридизованій р-орбіталі знаходиться неспарений електрон. Велика реакційна здатність радикалу •СН3 пояснюється прагненням неспареного електрона до утворення ковалентного σ-зв'язку (оскільки це дає виграш в енергії і стабілізує всю систему в цілому) та доступністю негібридизованої р-орбіталі для атаки.
Метильний радикал атакує молекулу хлору, при цьому відбувається гомолітичний розрив зв'язку в Cl2 і утворюється молекула хлорметану та новий радикал Хлору:
![]()
При вивченні процесів галогенування алканів з'ясувалося, що найскладніше відбувається одержання хлорметану СН3Cl, а утворення ди-, три- і тетрагалогенопохідних (СН2Cl2, CHCl3 i CCl4) проходить дуже швидко і супроводжується виділенням теплоти. Це пояснюється дією негативного індуктивного ефекту атома Хлору (див. § 5.2), завдяки якому відбувається поляризація σ-зв'язків С–Н, що значно полегшує їх розрив і подальше заміщення атомів Гідрогену в хлорметані СН3Cl атомами Хлору:

Радикал Хлору Cl• знов атакує молекулу СН4 і повторюються всі перелічені процеси. У міру накопичення в реакційній суміші першого продукту реакції – хлорметану СН3Сl, він стає об'єктом атаки вільним радикалом Cl•, тому поступово утворюються не тільки монохлоропохідні, а і ди-, три-, тетрахлоропохідні:
![]()
![]()
![]()
![]()
![]()
![]()
Процеси, що проходять подібним чином, називаються ланцюгові реакції, оскільки утворення в системі продуктів попереднього процесу автоматично ініціює протікання наступного.
Третя стадія – обрив ланцюгу відбувається при випадковому зіткненні двох вільних радикалів, внаслідок чого утворюється нейтральна молекула. Найчастіше обрив ланцюгу спостерігається при взаємодії двох радикалів Хлору, але можуть бути й інші варіанти, тому в реакціях галогенування алканів, крім бажаних речовин, утворюється велика кількість побічних продуктів, наприклад:
![]()
![]()
![]()
6.2.3 Реакції елімінування
Реакції елімінування Е (від англ. еlimination – усунення) – це процеси, при яких від молекули вихідної сполуки внаслідок розриву зв’язків відривається частинка, здатна до самостійного існування: молекула, іон, бірадикал (тобто радикал, що має два неспарені електроні). Залежно від взаємного розміщення атомів (Н і Х), що відщеплюються від вихідної молекули, розглядають типи елімінування:
- α-елімінування, коли частинка НХ відщеплюється від одного атома Карбону і утворюється нестабільний бірадикал карбен:
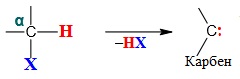
- β-елімінування – відщеплення протону Н+ і групи Х–від двох сусідніх атомів Карбону з утворенням кратного зв’язку:
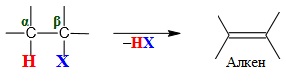
Як приклад β-елімінування можна навести реакцію дегідратація спирту – відщеплення води з утворенням алкенів:
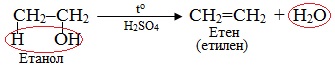
- [TEX]\gamma [/TEX]-елімінування, під час якого атоми Н і Х відриваються від двох атомів С, розділених однією чи декількома метиленовими групами (–СН2–), внаслідок чого утворюється циклічні вуглеводні. Наприклад, продуктом елімінування γ-заміщеної сполуки є тричленний цикл, що містить в головному ланцюгу три атоми С:
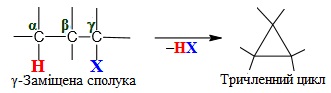
6.2.4 Реакції перегрупування
Реакції перегрупування – це хімічні процеси, в ході яких відбувається перехід (міграція) окремих атомів чи атомних груп від одних ділянок молекули до інших.
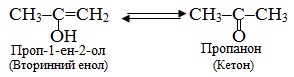
Як приклад можна навести кето-енольне перегрупування енолів.
Еноли (або заміщені вінілові спирти) – це ненасичені спирти, що містять сполучений з гідроксильною групою (ОН) sp2-гібридизований атом Карбону, який утворює подвійний зв’язок з сусіднім атомом С. В первинних енолах група ОН займає крайнє положення, а у вторинних знаходиться усередині ланцюгу:
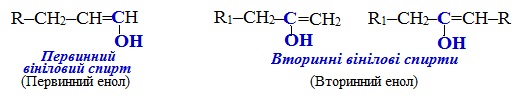
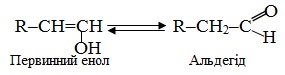
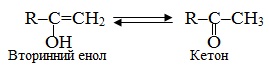
Еноли мають певну особливість: вони здатні ізомерізуватися відповідно до правило Ельтекова: Еноли піддаються швидкому перегрупуванню (кето-енольна таутомерія), перетворюючись в карбонільні сполуки: первинні вінілові спирти – в альдегіди, а вторинні – в кетони:
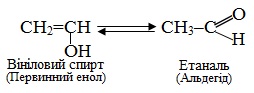
Наприклад, кето-енольне перегрупування вінілового спирту в етаналь (ацетальдегід) та проп-1-ен-2-олу – в пропанон (ацетон):
До реакцій перегрупування належить й ізомеризація алканів – процес, що супроводжується зміненням структури карбонового ланцюгу при збереженні вихідного складу вуглеводню. Шляхом ізомерізації з алканів нормальної будови одержують розгалужені ізомери, в яких метильний радикал (–СН3) найчастіше сполучений з другим від краю атомом С основного ланцюгу, наприклад:
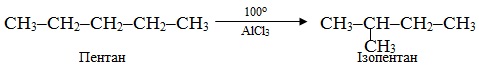
Ізомеризація характерна також для кумульованих дієнових вуглеводнів (в яких один атом С сполучається з сусідніми атомами Карбону двома подвійними зв’язками). Внаслідок ізомеризації утворюються алкіни, наприклад:

6.2.5 Реакції полімеризації
Реакції полімеризації певною мірою можна вважати різновидом реакцій сполучення.
Полімеризація – це процес послідовного приєднання молекул ненасиченої сполуки (мономер) одна до одної за рахунок розриву π-зв'язків і утворення нових σ-зв'язків між окремими елементарними ланками в макромолекулі високомолекулярної сполуки (полімер), яка при цьому утворюється.
Елементарна ланка – фрагмент структури, що повторюються в макромолекулі полімеру. Кількість елементарних ланок визначається кількістю молекул мономеру, які взяли участь у реакції полімеризації, і називається ступінь полімеризації n.
Під час полімеризації одна макромолекула високомолекулярної сполуки (скорочено – ВМС) може утворюватися з різної кількості молекул мономеру (n), тому полімер звичайно складається з макромолекул різної довжини і різної молекулярної маси.
Молекулярна маса полімеру – це середня величина, яку розраховують як добуток молярної маси мономеру на ступінь полімеризації за формулою
МВМС = n·Ммономеру.
Схематично процес полімеризації записують так:

До полімеризації здатні майже всі сполуки з подвійним зв'язком >C=C< в ланцюгу, які можна вважати похідними етену (тривіальна назва С2Н4 – етилен) з загальною формулою СН2=СН–Х, де Х – замісник. Наприклад, в акрилонітрилі, з якого одержують поліакрилонітрил (нітрон), роль такого замісника відіграє ціаногрупа –СN:

А в мономерах, при полімеризації яких отримують фторопласти (тефлон), замісниками є атоми галогенів, наприклад:

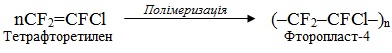
Іноді полімеризації піддаються суміші різних мономерів – такий різновид сумісної полімеризації називають сополімеризація, а одержану при цьому високомолекулярну сполуку – сополімером. Наприклад:
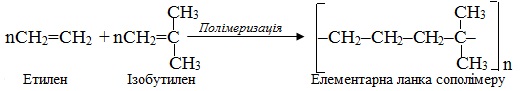
Процес сополімеризації особливо часто використовують для сумісної полімеризації алкадієнів з іншими ненасиченими сполуками: з вінілхлоридом СН2=СН–Сl, стиреном СН2=СН–С6Н5, акрилонітрилом СН2=СН–CN.
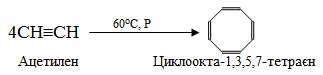
Скороченим варіантом полімеризації можна вважати процес олігомерізація – реакція сполучення двох чи декількох молекул мономеру, внаслідок чого утворюється ненасичений олігомер – продукт нормальної або циклічної будови. Залежно від умов (to, kat) олігомерація проходить різними шляхами, наприклад, для етину (тривіальна назва – ацетилен) можливі такі види олігомеризації:
- лінійна димеризація – сполучення двох молекул ацетилену:
![]()

- циклічна тетрамеризація, яка дає можливість одержувати ненасичені циклічні вуглеводні, що містять парну кількість (вісім чи більше) атомів Карбону в ланцюгу, наприклад, тетрамеризація етину С2Н2 (ацетилену):
Високомолекулярні сполуки інколи одержують й іншим шляхом – способом поліконденсації.
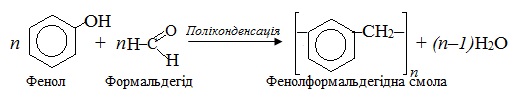
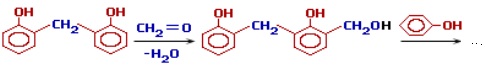
Поліконденсація – це реакція, при якій відбувається взаємодія молекул різних мономерів, внаслідок чого утворюються високомолекулярні сполуки і виділяються низькомолекулярні побічні продукти (H2O, NH3, HCl тощо). Наприклад, поліконденсація фенолу з метаналем (тривіальна назва – формальдегід), внаслідок якої одержують цінні продукти – феноформальдегідні смоли, або бакеліти:
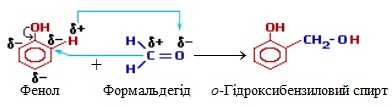
За механізмом перебігу ця реакція відносно фенолу проходить за типом електрофільного заміщення SE, а відносно формальдегіду – за типом нуклеофільного приєднання AN, як це видно на кольоровій схемі:
Гідроксибензиловий спирт, що утворився на першій стадії, вступає в подальшу взаємодію з фенолом, збільшуючи довжину ланцюгу:
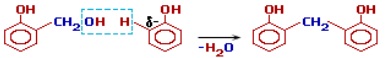
На наступному етапі відбувається приєднання ще однієї молекули формальдегіду і так далі:
6.3 Класифікація за валентним станом атомів Карбону
За зміною валентного стану атомів Карбону розглядають такі процеси:
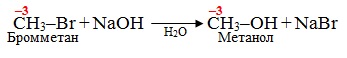
- Реакції невалентних перетворень, при яких ступінь окиснення атомів Карбону не змінюється. Необхідно зауважити, що таких реакцій в органічній хімії зовсім небагато, наприклад, реакція лужного гідролізу галогеноалканів:
- Окисно-відновні реакції, внаслідок яких відбувається змінення ступенів окиснення атомів Карбону. Звичайно при перебігу більшості органічних реакцій атом С (один чи декілька) змінюють ступінь окиснення Так, навіть в найпростішій з першого погляду реакції гідрування ненасиченого вуглеводню (приєднання водню Н2) спостерігається змінення ступенів окиснення двох атомів Карбону:
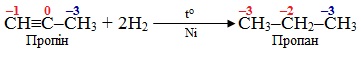
Окисно-відновні реакції за участю органічних сполук мають свої особливості порівняно з неорганічними ОВР, які в першу чергу пов’язані з визначенням ступенів окиснення атомів С. При цьому розглядають атом Карбону тільки в тому фрагменті молекули, який піддається перетворенню, не звертаючи уваги на інші. Друга особливість: вважається, що сума ступенів окиснення всіх атомів у даному фрагменті дорівнює нулю; по-третє, атомам інших елементів приписують такі ступені окиснення: H+1, O–2, N–3, S–2, P–3. Рівняння електронного балансу складають таким же чином, що і в неорганічних реакціях. Наприклад, окислення стирену калій перманганатом у сульфатнокислому середовищі:
![]()
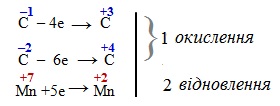
6.4 Класифікація за природою реагентів
Залежно від природи частинок, що приєднуються до органічної сполуки, чи відщеплюються від неї, органічні реакції поділяють на певні типи. Назва окремого типу реакцій приєднання походить від латинської чи грецької назви частинки, що приєднується до субстрату, а до назви реакцій відщеплення цієї ж частинки додають префікс де-. Наприклад, приєднання Н2О називається гідратація (від слова «гідро» – вода), а відщеплення Н2О – дегідратація.
1. Гідрування і дегідрування – відповідно це реакції приєднання водню і зворотний процес – відщеплення Н2. Обидва процеси, як правило, проводять в жорстких умовах (температура, тиск, каталізатор). Гідруванню піддають різні групи сполук, в молекулах яких містяться кратні зв’язки:
![]()
Щоб оцінити здатність алкенів різної будови до приєднання Н2, корисно брати до уваги правило Лебедєва: водень тим легше приєднується до кратного зв’язку, чим менше замісників міститься біля нього. Отже, можна прогнозувати, що пропен нормальної структури легше приєднує Н2 порівняно з розгалуженим 2-метилпропеном:
![]()
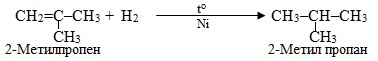
Алкіни, незважаючи на більшу ненасиченість, піддаються гідруванню важче, ніж алкени, причому реакція проходить у два етапи:
![]()
Якщо вуглеводень містить одночасно подвійні та потрійні зв’язки, то водень спочатку завжди приєднується до подвійного зв’язку, а потім – до потрійного, наприклад:
![]()
Реакції дегідрування приводять до утворення ненасичених аліфатичних чи навіть до циклічних сполук, як показано на прикладі бутану:
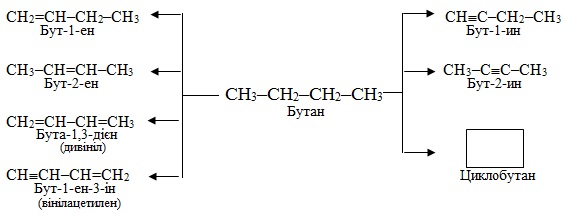
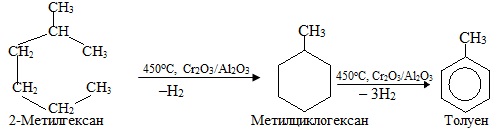
Реакція відщеплення водню від алканів, що містять в ланцюгу 6-9 атомів С, називається ароматизація, під час якої відбувається не тільки дегідрування насичених вуглеводнів, але і процеси циклізації та ізомеризації, в результаті чого одержують ароматичні вуглеводні, наприклад:
2. Галогенування і дегалогенування – це відповідно введення атомів галогенів Hal в органічні сполуки і відщеплення їх. Продуктами реакцій галогенування є ди- чи полігалогенопохідні – залежно від кількості приєднаних атомів галогену, наприклад:
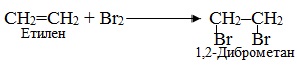
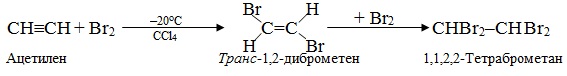
Реакції дегалогенування дають різні продукти залежно від складу і будови вихідного галогеновуглеводня і природи реагенту, наприклад, при дії цинкового пилу або магнієвих ошурків на дигалогеналкани одержують ненасичені аліфатичні чи цикліклічні вуглеводні, наприклад:
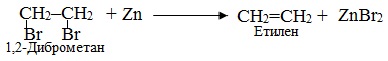
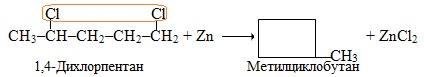
Окремим випадком реакцій дегалогенування є реакція Вюрця, що проходить при взаємодії галогенопохідного з металічним натрієм при незначному нагріванні. При цьому утворюється алкан, який має вдвічі довший ланцюг, ніж вихідний галогеналкан:
![]()
Проте слід брати до уваги, що при дії натрієм на суміш двох різних галогенопохідних реакція одночасно перебігає за трьома різними схемами з утворенням суміші трьох алканів, як показано на схемі:
![]()
![]()
![]()
3. Гідрогалогенування і дегідрогалогенування – це відповідно реакції приєднання гідрогенгалогенідів HHal і відщеплення гідрогенгалогенідів. Гідрогенгалогеніди приєднуються до ненасичених сполук, що містять кратні (подвійні чи потрійні) зв’язки з утворенням галогенопохідних.
Якщо вихідний субстрат (алкен, алкін чи карбоциклічний вуглеводень) має несиметричну будову, то напрямок приєднання до нього гідрогенгалогенідів HHal (а також інших реагентів, що складаються з полярних молекул: H2O, HNH2), визначається відповідно до правило Марковникова (див. § 6.2.1).
Необхідно зауважити, що не всі реакції приєднання гідрогенгалогенідів HHal та інших полярних молекул типу НХ до кратних зв’язків підлягають правилу Марковникова. Якщо у β-положенні відносно кратного зв’язку міститься сильна електроноакцепторна група (наприклад, –СHal3, –СООН, –СНО, –NO2, –С≡N), то приєднання HHal проходить всупереч правилу Марковникова. Це зумовлюється тим, що електроноакцепторна група за рахунок негативного індуктивного (–I) чи мезомерного (–M) ефекту відтягує в свій бік електронну густину подвійного зв’язку, а це призводить до його поляризації та виникненню часткових зарядів δ+ і δ– на атомах Карбону, як це показано на схемі:
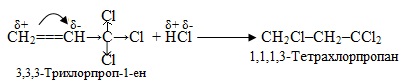

Ще одним виключенням з правила Марковникова є реакція приєднання HBr в присутності пероксидних сполук, які викликають пероксидний ефект Харраша (див. § 6.2.1) і направляють перебіг реакції всупереч правилу Марковникова, наприклад:
![]()
Реакції дегідрогалогенування – відщеплення атомів Н і Hal від двох сусідніх атомів Карбону – проводиться при дії на галогенопохідні спиртовим розчином лугу. У випадку несиметричних галогеновуглеводнів реакція відбувається згідно з закономірністю, відомою як правило Зайцева: У реакціях дегідрогалогенування атом Гідрогену відщеплюється переважно від найменш гідрогенізованого атома Карбону, сусіднього відносно зв’язку С–Наl, наприклад:

4. Гідратація і дегідратація – це відповідно реакції каталітичного приєднання молекул води і відщеплення молекул H2O від органічних сполук, до складу яких входить гідроксильна група ОН.
Гідратація несиметричних ненасичених сполук проходить згідно з правилом Марковникова, оскільки молекула води є полярною, наприклад:
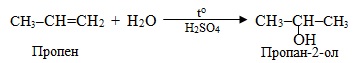
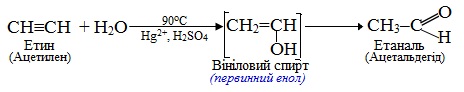
Гідратація сполук, що містять потрійний зв’язок, називається реакція Кучерова; вона відбувається при каталітичній дії солей меркурію (ІІ) і сульфатної кислоти і приводить до утворення кетонів, за єдиним виключенням: продуктом приєднання води до етину (ацетилену) є альдегід. Реакція проходить через проміжну стадію утворення нестійких енолів, які швидко перетворюються в карбонільні сполуки внаслідок кето-енольній таутомерії (див. § 6.2.4), наприклад:
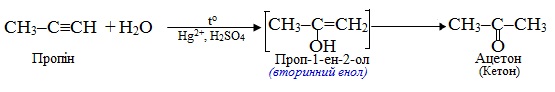
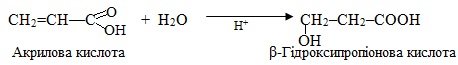
Гідратація ненасичених сполук, молекули яких містять в β-положенні відносно кратного зв’язку сильну електроноакцепторну групу (–СHal3, –СООН, –СНО, –NO2, –С≡N), відбувається всупереч правилу Марковникова:
При дегідратації несиметричних сполук, що містять групу ОН, діє правило Зайцева: разом з групою ОН одночасно відбувається відщеплення атома Н від наймеш гідрогенізованого атома С, сусіднього відносно зв’язку СН–ОН:
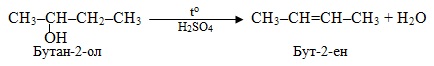
5. Нітрування – введення нітрогрупи (–NO2) в сполуку при дії нітратної кислоти. Нітрування аліфатичних, аліциклічних і ароматичних вуглеводнів традиційно називають реакція Коновалова, наприклад:
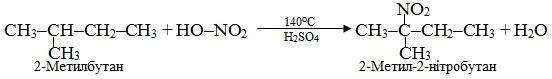
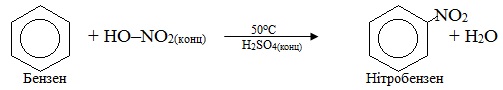
Внаслідок реакцій нітрування утворюються нітросполуки, які не слід плутати з естерами нітратної кислоти. На відміну від нітросполук в молекулах нітратних естерів нітрогрупа NO2 сполучена з вуглеводневим радикалом через атом Оксигену, як це видно з порівняння загальних формул:
![]()
6. Сульфування – реакція введення сульфогрупи (–SO3H) в молекулу органічної сполуки при дії на неї концентрованою сульфатною кислотою або олеумом (розчин SO3 в концентрованій H2SO4). Внаслідок реакції сульфування утворюються сульфокислоти (інша назва – сульфонові кислот), наприклад:
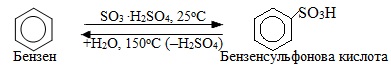
Десульфування – відщеплення сульфогрупи проходить при гідролізі сульфонових кислот гострою парою:
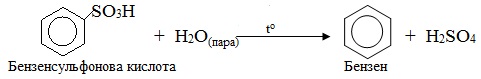
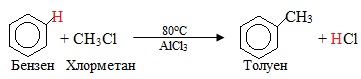
7. Алкілювання – введення в молекулу органічної сполуки вуглеводневого замісника – алкільного радикалу (–CnH2n+1), в тому числі метилювання – введення в молекулу метильної групи (–СН3). Алкілювання може проходити як по атому Карбону, так і по гетероатомам. Залежно від цього розглядають С-, О- N-, O-, S-алкілювання.
С-алкілювання розглянемо на прикладі бензену. Введення алкільного радикалу (СnH2n+1) в бензенове кільце в присутності каталізатора називають – реакція Фріделя-Крафтса. Продуктами алкілювання є гомологи бензену. В якості алкілювального реагенту найчастіше використовують галогеналкани СnH2n+1Hal чи алкени СnH2n, наприклад:
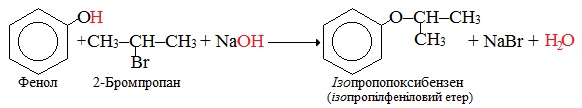
О-Алкілювання – заміщення атома Гідрогену гідроксильної групи, внаслідок чого алкільний замісник сполучається з атомом Оксигену. При цьому утворюються змішані етери, наприклад:
N-алкілювання – заміщення атома Гідрогену аміногрупи, внаслідок чого алкільний замісник сполучається з основним ланцюгом через атом N, наприклад:
![]()
8. Ацилювання – реакція введення ацильного залишку (R–CO–), який утворюється при відщепленні гідроксильної групи від карбонової кислоти. Якщо ацилом є залишок мурашиної кислоти (форміл HCO–), то процес називають формілювання, якщо вводять залишок оцтової кислоти (ацетил CH3CO–) – ацетилювання. Залежно від реакційної здатності субстрату і реагенту в якості ацилювальних реагентів вибирають карбонові кислоти чи їх функціональні похідні, активність яких в реакціях ацилювання зменшується в такій послідовності:
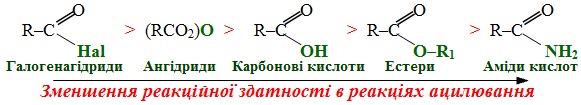
Подібно до алкілювання введення ацильного радикалу теж може проходити не тільки по атому Карбону, але і по гетероатомам, залежно від чого реакцію поділяють на окремі типи: С-, О- N-, O-, S-ацилювання.
С-ацилювання ненасичених і ароматичних вуглеводнів приводить до утворення кетонів, наприклад:
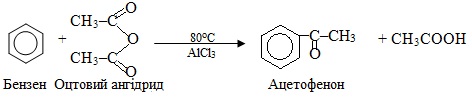
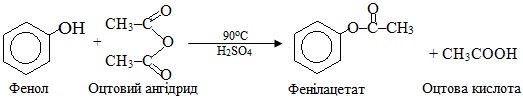
О-Ацилювання – заміщення атома Гідрогену в гідроксилі на ацильну групу RC(O)–, при цьому утворюються естери, наприклад:
N-Ацилювання – заміщення атома Гідрогену в аміногрупі NН2 ацильним залишком RC(O)–. Ацилювання амінів проводять карбоновими кислотами та їх похідними, внаслідок якого первинні та вторинні аміни перетворюються відповідно у моно- і дизаміщені аміди кислот, наприклад, ацилювання оцтовою кислотою:
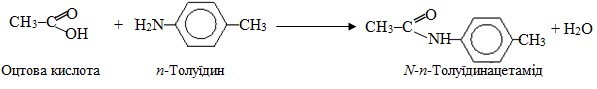
Подібним чином проходять реакції N-ацилювання амінів з галогенангідридами і ангідридами карбонових кислот відповідно до загальної схеми:
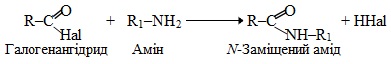
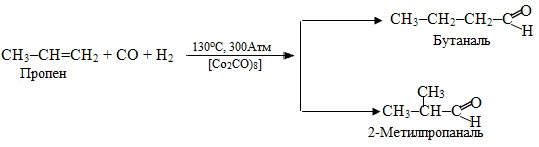
9. Карбонілювання – найчастіше це взаємодія ненасичених вуглеводнів із сумішшю СО + Н2, внаслідок чого утворюються альдегіди чи кетони, що містять оксогрупу (–СН=О чи >C=O). Для реакцій карбонілювання інколи вживають й інші назви – оксосинтез, гідроформілювання.
Внаслідок карбонілювання алкену одержують, як правило, суміш альдегідів (залежно від того, до якого атома Карбону при подвійному зв'язку приєднується СО і Н2), однак нерозгалужений альдегід отримується у переважній кількості, наприклад:
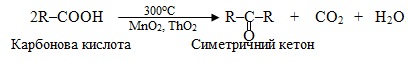
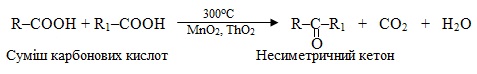
10. Карбоксилювання – введення карбоксильної групи –СООН при взаємодії ненасичених вуглеводнів з сумішшю СО + Н2О (або СО2 + Н2), і декарбоксилювання – відщеплення від карбонових кислот та їх похідних карбоксильної групи –СООН чи просто –СОО–, тобто карбон (ІV) оксиду СО2.
Звичайно карбоксилюванню піддають алкени і алкіни, внаслідок чого утворюються монокарбонові кислоти, що містять на один атом Карбону більше, ніж у вихідному вуглеводні, наприклад:
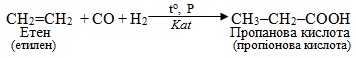
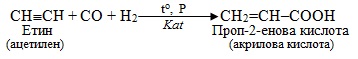
Реакції декарбоксилювання мають важливе значення в органічному синтезі, оскільки завдяки їм одержують продукти, що належать до різних класів сполук. Відомо багато способів декарбоксилювання вихідних речовин, які містять карбоксильні групи. Розглянемо окремий випадок – декарбоксилювання солей за реакція Дюма, під час якої відбувається реакція лужного плавлення карбоксилатів (тобто солей лужних металів). На схемі для наочності виділено склад побічного продукту – натрій карбонату:
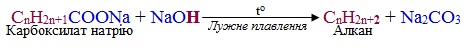
Декарбоксилюванню за реакцією Дюма піддають не тільки солі (СnH2n+1–COOMe), але і нерозчинні карбонові кислоти (СnH2n+1–COOH, особливо при n ≥ 6):
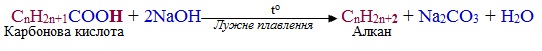
Ще один метод декарбоксилювання – реакція Кольбе, яку проводять при електролізі водних розчинів солей (карбоксилатів) лужних металів. При цьому на аноді внаслідок окислення кислотного залишку утворюються вільні алкільні радикали (•СnH2n+1), які завдяки наявності неспареного електрона мають підвищену реакційну здатність і дуже швидко сполучаються один з одним в молекулу алкану з більш довгим ланцюгом:
![]()
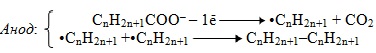
![]()
Декарбоксилювання за методом суха перегонка солей, для якого найчастіше використовують солі магнію чи лужноземельних металів. У наведеному прикладі жовтим колом обведений склад побічного продукту – карбонатної солі:
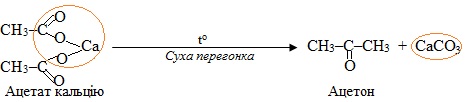
Якщо сухій перегонці піддають суміш двох різних солей, то одержують змішаний кетон, що містить вуглеводневі радикали обох солей, наприклад:
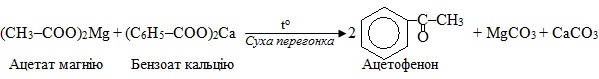
В тому випадку, коли одним з компонентів вихідної суміші є сіль метанової (мурашиної) кислоти, одержують не кетони, а альдегіди, наприклад:
![]()
Декарбоксилювання карбонових кислот дає можливість одержати продукти різної будови – залежно від складу і структури вихідних кислот:
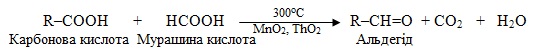
Дикарбонові кислоти при декарбоксилювання дають циклічні кетони:
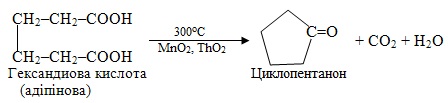
6.5 Приклади розв'язання типових задач
Приклад 6.1. Встановіть, які продукти переважно утворюються внаслідок монобромування 2,4-диметилгексану. За яким механізмом проходить ця реакція?
Розв’язок. Розгалужені алкани з великою довжиною ланцюгу вступають в реакції бромування селективно (вибірним чином) – переважно біля третинного атома Карбону. Такому напрямку реакції сприяє позитивний індуктивний ефект +І метильних радикалів (–CН3), які поляризують і послаблюють зв’язки С–Н саме у третинного атома Карбону. Тому при монобромуванні 2,4-диметилгексану, що містить два третинних атоми С, з рівною вірогідністю утворюватимуться два ізомерні галогеналкани:
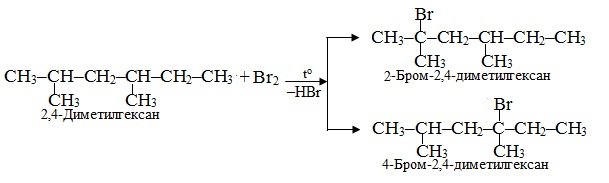
Реакція проходить за механізмом радикального заміщення SR, найбільш характерного для насичених вуглеводнів.
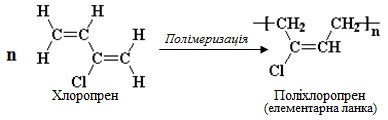
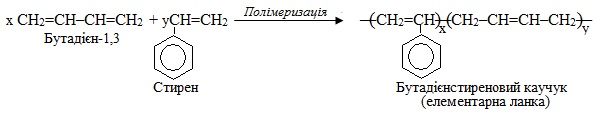
Приклад 6.2. Скласти схеми: а) полімеризації 2-хлорбута-1,3-дієну; б) сополімеризації бута-1,3-дієну і стирену. Назвати продукти.
Розв’язок. а) При полімеризації 2-хлорбута-1,3-дієну (тривіальна назва – хлоропрен) утворюється полімер, відомий під назвою хлоропреновий каучук. Схема полімеризації:
б) Продуктом сополімеризації бута-1,3-дієну і стирену є сополімер, який називають бутадієнстиреновий каучук. Схема сополімеризації:
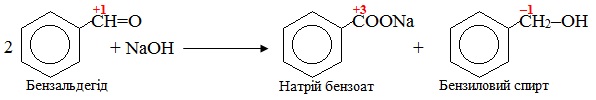
Приклад 6.3. При дії лугом на альдегід, який в α-положенні не містить жодного атома Н, утворюються два продукти – спирт і карбоксилат (сіль карбонової кислоти). Скласти схему відповідної реакції, що проходить при взаємодії бензальдегіду з NaOH. Чи належить вона до окисно-відновних реакцій?
Розв’язок. Задана реакція є окисно-відновною, оскільки при її перебігу відбувається змінення ступеню окиснення бензильного атома С, безпосередньо сполученого з бензеновим кільцем. Реакція проходить за типом диспропорціонування, тому ступінь окиснення змінюється тільки у одного атома Карбону, як це видно з схеми реакції:
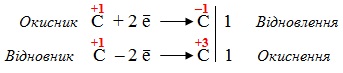
Приклад 6.4. Визначити, до яких типів за природою частинок, що приєднуються чи відщеплюються, належать реакції:
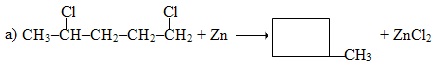
![]()
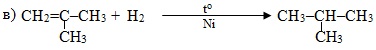
![]()
![]()
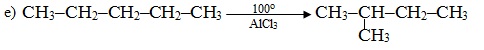
Розв’язок. а) При нагріванні з цинковим порошком ізольованих дигалогеналканів, в яких атоми С, що сполучені з атомами галогену, відокремлені один від одного двома чи більше метиленовими групами (СН2), утворюються циклоалкани. Під час реакції від вихідної сполуки відщеплюються два атоми Hal, реакція є реакцією дегалогенування:
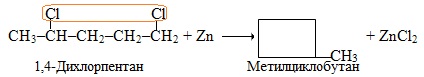
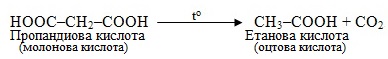
б) При нагріванні дикарбонової молонової кислоти, що містить в ланцюгу три атоми С, відбувається відщеплення однієї карбоксильної групи (СОО), тому це реакція декарбоксилювання:
в) Приєднання водню до ненасиченого вуглеводню за відповідних умов – це реакція гідрування:
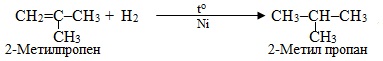
г) При дії спиртовим розчином на моногалогеналкани проходить реакція дегідрогалогенування, оскільки відбувається відщеплення атомів Н і Br, сполучених з двома сусідніми атомами С:
![]()
д) При нагріванні гідроксикислот відщеплюються атом Н і група ОН від двох сусідніх атомів С. Отже, проходить реакція дегідратації:
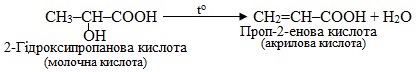
е) При нагріванні алкану лінійної будови в присутності каталізатора AlCl3 відбувається реакція ізомеризації:
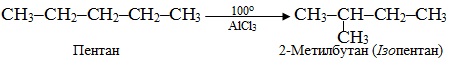
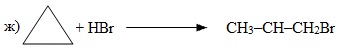
ж) Малі цикли, що містять 3-4 атоми С в кільці, здатні розривати цикл і приєднувати галогеноводні, тому дана реакція належить до реакцій гідрогалогенування:
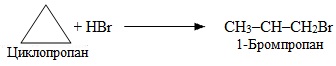
Приклад 6.5 Скласти рівняння реакції, що проходить при ароматизації вуглеводню заданої структури:
![]()
Розв’язок. Ароматизація – це утворення ароматичних вуглеводів внаслідок одночасного дегідрування (–Н2) і циклізації аліфатичних вуглеводнів, що містять 6-9 атомів С в молекулі. Оскільки в насичених вуглеводнях атоми Карбону перебувають в sp3-гібридизованому стані, який зумовлює розмір валентного кута 109,5о, головний ланцюг може набувати зигзагоподібної чи клешневидої форми, що сприяє просторовому зближенню атомів С1 і С6. З урахуванням цього зобразимо просторову структурну формулу заданої сполуки:
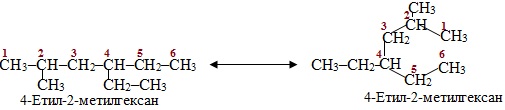
Така особливість будови 4-етил-2-метилгексану спрощує за певних технологічних умов відщеплення атомів Гідрогену від С1 і С6 і виникненню нового зв’язку С1–С6. Перебіг реакції ароматизації відбувається за схемою:

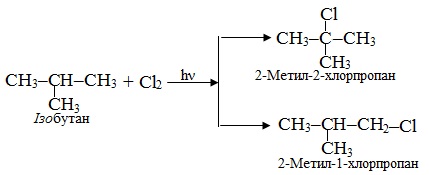
Приклад 6.6. Скласти схеми реакцій хлорування і нітрування ізобутану.
Розв’язок. На відміну від брому, який виявляє схильність до селективного заміщення переважно біля третинного чи вторинного атома С, більш активний хлор з рівною вірогідністю може атакувати атоми С будь-якої природи, тому внаслідок реакції утворюється суміш галогенопохідних. Реакція радикального заміщення проходить при опромінюванні розсіяним світлом (hν) за схемою:
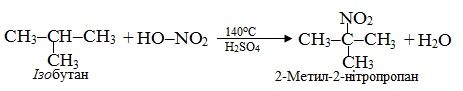
Нітрування – це реакція Коновалова, яка проходить майже завжди селективно, тому внаслідок неї отримують головним чином третинну нітросполуку:
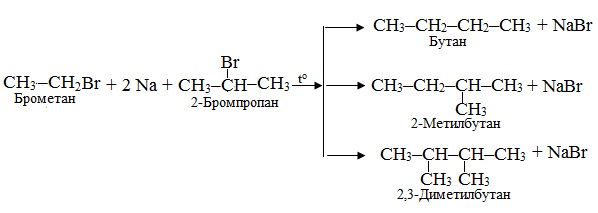
Приклад 6.7. Визначити склад продуктів, що утворюються при незначному нагріванні металічного натрію з сумішшю брометану і 2-бромпропану.
Розв’язок. При нагріванні галогеналканів з металічним натрієм відбувається реакція Вюрця. У випадку суміші двох різних галогенопохідних одержують продукти, що містять три різних алкани.
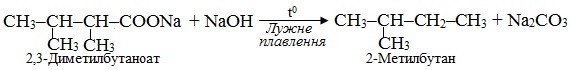
Приклад 6.8. Встановити, яку сіль піддали реакції лужного плавлення, якщо продуктом є 2-метилбутан.
Розв’язок. Лужне плавлення солей (карбоксилатів) – це процес декарбоксилювання за реакцією Дюма. Для одержання внаслідок такої реакції 2-метилбутану в якості вихідних карбоксилатів можна взяти одну з двох солей: 2,3-диметилбутаноат натрію і 2-метилпентаноат натрію. Реакції проходять відповідно до схем:
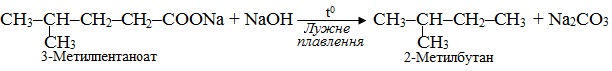
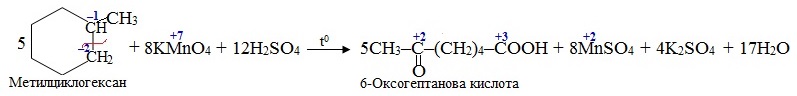
Приклад 6.9. Cкласти рівняння реакції окислення метилциклогексану розчином калій перманганату, що підкислений сульфатною кислотою.
Розв’язок. Окислення циклоалканів супроводжується руйнуванням замкнутого карбонового ланцюгу, причому, розрив зв’язку С–С відбувається в місці, наближеному до бокового радикалу –СН3. З цього випливає, що продукт окислення буде містити в ланцюгу вже не шість, а сім атомів С. Отже, за місцем розриву зв’язку С–С окислюватися будуть обидва атоми Карбону (С1 і С6). Перший у циклі атом С (сполучений з радикалом СН3) перебуває в ступені окиснення –1, а окислюється до ступеня окиснення +2; в новому ланцюгу він буде другим від краю, тому перетвориться на карбонільний (>С=О). Шостий у циклі атом С, що має ступень окиснення –2, після реакції набуває ступеня окиснення +3 і буде знаходитися з краю нового ланцюгу, тому перетворюються на карбоксильний (СООН). Реакція проходить відповідно до рівняння:
Коефіцієнти в рівнянні реакції проставлення на підставі електронного балансу:
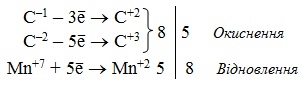
Приклад 6.10. Написати рівняння реакції, що відбувається при дії цинковим порошком на 1,5-дибром-2-метилциклопропан.
Розв’язок. При дії цинковим порошком на ізольовані дигалогеналкани утворюються циклоалкани. Розмір цикла визначається відстанню між атомами галогену. Оскільки в вихідній сполуці атоми брому сполучені з першим і п’ятим атомами С, то внаслідок реакції утворюється п’ятичленний цикл: