-
Синдром Марфана
- Фенілкетонурія
-
Гомоцистинурія
-
Муковісцидоз (кістозний фіброз)
- Вроджений синдром гіпотиреозу
- Нейрофіброматоз
-
Синдром Елерса-Данлоса
Ключові терміни:
(Classical), (Hypermobility), (Vascular), ), ,, , , 130000, 130010, 130020, 130050, 130060, 2, 225400, 225410, 229200, 7A, AD, B, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, OMIM, PLOD1, TNXB, `, І, Артрохалаз, Аутосомно-домінантний дефект, що викликає недолік ферменту, званого Лізин гідролазами. Нечасто зустрічається, Вражає 1 людини на 10 000 - 15 000, викликаний аутосомн, Вражає колаген типу I. Вкрай рідкісний, описано всього близько 30 випадків., Вражає приблизно 1 людину на 100000. Викликаний аутосомно-домінантним дефектом у синтезі колагену типу III, Вражає приблизно від 2 до 5 осіб на 100 000. Зачіпає колаген типу V, також колаген типу I., Г, Ген(, Дерматоспараксис (Dermatosparaxis), К, Клас, Номер, Опис, С, Синдром Елерса-Данлоса, Також вкрай рідкісний, описано близько 10 випадків., з дефіцитом тенасціна-X, відповідно. Виявляється в гіперрухливості суглобів; ураження шкіри виражене менше. Характерні хронічні м'язово-скелетні болі., и, ичний, м, механізмом. Виникає в результаті мутації будь-якого з двох генів, які викликають Судинний тип і, о-домінантним, оз (Kyphoscoliosis), описано трохи більше 60 випадків., пер, рухливість, тип, тип 1, тип 3, тип 4, тип 6, тип 7C, удинний, фоскол, я, я (Arthrochalasis), іТема 2. ЗАГАЛЬНА ХАРАКТЕРИСТИКА МОНОГЕННОЇ ПАТОЛОГІЇ. КЛІНІКА І ГЕНЕТИКА ОКРЕМИХ ФОРМ МОНОГЕННИХ ХВОРОБ.
Моногенні захворювання - розлади, спричинені успадкуванням одного дефектного гена, відомі як моногенні захворювання або порушення одного гена.
Існує понад 6000 відомих одногеневих розладів, які виникають у 1: 200 новонародженних.
Деякими прикладами є муковісцидоз, серповидна клітинна анемія, синдром Марфана, хвороба Хантінгтона, спадковий гемохроматоз, м'язова дистрофія Дюшенна, гемофілія та хвороба Тея-Сакса.
Одно-генні або моногенні захворювання можна розділити на три основні категорії: аутосомно-домінантний, аутосомно-рецесивний і Х-зв'язаний.
Синдром Марфана
Синдром Марфана є аутосомно-домінантним розладом (25% випадків - нові мутації), що впливають на сполучні тканини, викликані мутацією в гені, який кодує фібрилін 1 (FBN1). Фібрилін є основним компонентом мікрофібрил, що дозволяє тканинам розтягуватися неодноразово без ослаблення. Оскільки фібрилін пацієнта є ненормальним, його сполучна тканина є меншою, ніж звичайна, що послаблює або пошкоджує підтримуючі структури всього тіла.
Антуан Марфан (1858-1942) вперше описав його в 1896 році. Синдром Марфана впливає на три основні органи організму: серце і систему кровообігу, кістки та м'язи, а також очі.
Частота захворювання становить принаймні 1 з 10000.
Ознаки та симптоми
Опорно-рухова система (рис.1):
• високий зріст
• довгі кінцівки
• арахнодактилія кистей та стоп
• астенічна конституція
•вузький лицевий череп з «пташиним» виразом обличчя
• вузька та деформована воронкоподібна або кулеподібна грудна клітка
• кіфосколіотична деформація хребта
• гіпермобільність суглобів та сухожилок
• плоскостопість
• довге вузьке обличчя з глибокими очима
• високе вузьке піднебіння та скупчення зубів
• спондиліолітіоз
• екстракція суглобів
• pectus excavatum (грудина пацієнта запала всередину)
• pectus carinatum (грудна кістка випирає назовні і звужується).
• виступаюча вертикальна частина (вертлюжна оболонка стає більш глибокою, ніж звичайно під час росту)
Система зору:
• ектопія лентис (підвивих кришталика)
• міопія (короткозорість)
• відшарування сітківки (може призвести до сліпоти)
• глаукома
• катаракта
• блакитні склери
• гіперметропія високого ступеню
• аніридія
• афакія
• колобома
Розлади з боку ЦНС:
• анізокорія
• ністагм
• асиметрія сухожилкових рефлексів
• пірамідні розлади
Серцево-судинна система:
Близько 90% хворих на синдром Марфана мають серцеві ускладнення.
• порушення внутрішньошлуночкової провідності
• помірні ознаки гіпертрофії міокарду лівого шлуночка та передсердя
• зміни у магістральних судинах
• аортальна недостатність
• пролапс мітрального клапану
• порушення внутрішньосерцевої гемодинаміки
• мітральна недостатність, яка пов`язана з розвитком міксоматозної дегенерації стулок,збільшенням їх площини, розширенням фіброзного кільця, подовженням хорд, «розбовтаністю» стулок та збільшенням пролабірування.
• аортальна регургітація
• інфекційний ендокардит
• розширення кореня аорти та легеневої аорти
• аневризми синусів Вальсальви
• «висяче», «крапельне» серце
Інші проблеми:
• спонтанний пневмоторакс
• обструктивний апное уві сні
• емфізема
• striae (розтяжки шкіри)
• грижі
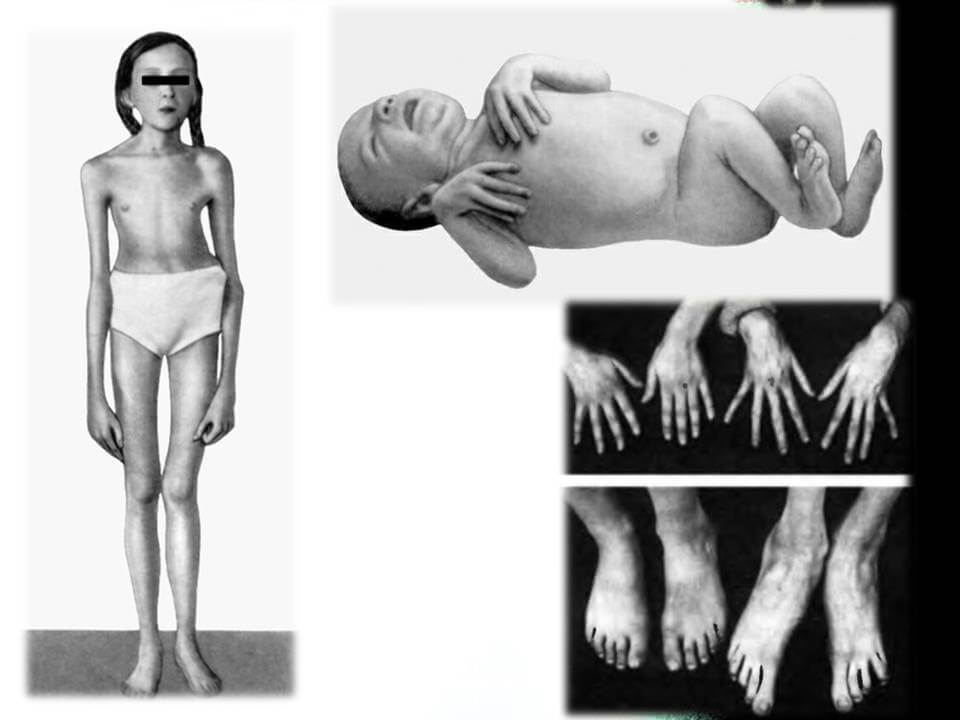
Рис. 1 - Синдром Марфана
Діагностика
Діагноз синдрому Марфана ставиться на основі сімейного анамнезу, вираженості симптоматики, фізикального огляду, результатів функціонального, рентгенологічного, офтальмологічного і генетичного досліджень, ЕКГ і ЕхоКГ і лабораторних досліджень.
Під час діагностування враховуються фенотипичні діагностичні тести, які визначають співвідношення кисті та зростання, довжину середнього пальця, індекс статури Варги, тест великого пальця на арахнодактилію, тест охоплення зап'ястя.
ЕКГ визначає порушення ритму серця, виражену гіпертрофію міокарда.
ЕхоКГ виявляє дилатацію аорти, клапанну регургитацию, пролапс мітрального клапана, збільшення розмірів лівого шлуночка, розриви хорд.
Рентгенографія грудної клітини дозволяє побачити розширення дуги аорти і кореня, збільшення розмірів серця; рентгенографія кульшових суглобів показує протрузію вертлюжної западини
КТ і МРТ серця і судин роблять для того, щоб виявити дилатацію і аневризму аорти; МРТ хребта показує ектазія твердої мозкової оболонки.
Аортографія проводиться при підозрі на аневризму і розшарування аорти.
Біомікроскопія і офтальмоскопия показують ектопію кришталика.
Генетична ідентифікація показує зміни (мутації) в гені FBN1.
У разі синдрома Марфана вдаються до диференціальної діагностики з хворобами, схожими з синдромом, до них відносяться: гомоцистинурія, вроджена контрактурна арахнодактилія (синдром Білса), спадкова артроофтальмопатія (синдром Стіклера), MASS-синдром, синдром Елерса-Данлоса, Лойса-Дитца, Шпрінцена- Голдберга, сімейна ектопія кришталика і ін.
Лікування
До терапії синдрому Марфана відноситься консервативна і хірургічна корекція серцево-судинних порушень, уражень скелета і органи зору.
Лікування, перш за все, спрямоване на профілактику розвитку захворювання і ускладнень в серцево-судинній системі. Якщо діаметр аорти до 4 см хворому призначають антагоністи кальцію, β-адреноблокатори (пропанолол, атенолол) або інгібітори АПФ. Хірургічне втручання проводиться тільки, коли діаметр аорти становить більше 5 см при пролапсі мітрального клапана, недостатності клапанів серця, висхідної частини і розшаруванні аорти.
При синдромі Марфана виконують реконструктивні операції на аорті. У разі необхідності виконується протезування мітрального клапана. Хворим з синдромом Марфана призначається корекція зору методом підбору окулярів і контактних лінз, а в складних випадках - шляхом лазерного або хірургічного лікування хвороб зору. Дітям зі скелетними порушеннями проводять хірургічну стабілізацію хребта, ендопротезування кульшових суглобів, торакопластику.
Хірургічне лікування сколіозу передбачає випрямлення хребта металевими стрижнями і спресування хребців у випрямленому положенні.
Спондиліолістезію лікують фіксатором у легких випадках. Якщо прослизання перевищує 30 градусів, зруйнований хребет може вимагати хірургічного перебудови.
Pectus excavatum і pectus carinatum можна лікувати хірургічним шляхом. У pectus excavatum деформована грудина та ребра піднімаються і випрямляються металевим стрижнем. Через чотири-шість місяців видаляється в амбулаторних умовах.
Біль у ногах або кінцівках звичайно обробляють м'яким анальгетиком (ацетамінофеном). Пацієнтам із синдромом Марфана слід подумати про носіння взуття на низьких підборах, спеціальних подушках або ортопедичних вставках.
Пацієнтам з синдромом Марфана необхідно провести ретельне обстеження очей, в тому числі випробування на щілинній лампі, для тестування на дислокацію об'єктива та короткозорості. Дислокацію можна лікувати комбінацією спеціальних окулярів та щоденним використанням офтальмологічних крапель 1% атропіну сульфату або хірургічним шляхом. Глаукому можна лікувати ліками або хірургічним шляхом. Катаракти стають все більш успішними при хірургії імплантату. Усім особам із синдромом Марфана слід вчити визнати ознаки відшарування сітківки (раптове розмивання зору в одному оці поступово погіршується без болю чи почервоніння).
Діти з синдромом Марфана повинні оглядатися стоматологами при кожному обстеженні для профілактики скупчення зубів і можливих невідповідностей, і при необхідності звернутися до ортодонта.
Людям із синдромом Марфана слід уникати спорту або професій, які потребують важкої ваги, грубого фізичного навантаження або швидких змін атмосферного тиску (наприклад, підводного плавання).
Однак регулярні дозовані фізичні вправи корисні для пацієнтів. Хорошим вибіром буде жваву ходьба, повільний темп бігу.
Фенілкетонурія
Фенілкетонурія (ФКУ) або фенілпіровиноградна олігофренія являє собою успадкований, аутосомно-рецессивний розлад, що характеризується дефіцитом ферменту фенілаланін-4-гідроксилази, який перетворює в тирозин фенілаланін. Це сприяє накопиченню в рідинах і тканинах організму хворої дитини фенілаланіну і його похідних (фенілмолочна, фенілпировиноградна і фенілоцтова кислоти, фенілетиламин, фенілацетилглютамін та ін). Вони надають токсичну дію на центральну нервову систему, сприяють порушення білкового обміну, а також метаболізму гормонів, гліко - і ліпопротеїдів.
Частота виникнення ФКУ становить 1 на 10000 народжених, але захворюваність у кавказьких та індіанських популяціях вище, ніж у афроамериканських, латиноамериканських та азіатських країнах. Майже з однаковою частотою захворювання зустрічається у хлопчиків та дівчаток.
Одним з найважчих наслідків захворювання є ураження головного мозку і подальше порушення розвитку дітей, як психічного, так і фізичного. Крім того, виникають розлади рухів і м'язового тонусу. Вперше класичну фенілкетонурію в 1934 році описав А. Феллинг.
Клінічні ознаки
Дитина з фенілкетонурією відразу ж після народження має цілком здоровий вигляд. Перші симптоми захворювання починають проявлятися у віці 2-х - 6-ти місяців. Такі діти бувають дуже млявими, дратівливими, неспокійними, їх не цікавить навколишній світ, можливе блювання. Психічна затримка розвивається поступово, і вона може не бути очевидною протягом перших кількох місяців. Інтелект пацієнтів з ФКУ, як правило, нижче, ніж IQ їх здорових однолітків.
Після 6 місяців такі діти значно відстають у психічному розвитку (близько 10% страждають слабким ступенем олігофренії, а близько 60% - ідіотією). Вони нормального або низького росту. Пізно починають прорізуватися зуби, трохи зменшуються розміри черепа, такі діти починають сидіти і ходити дуже пізно.
Для дітей з фенілкетонурією характерна своєрідна хода: вони ходять, гойдаючись, невеликими кроками, стоять, розставивши ноги, які зігнуті в тазостегнових і колінних суглобах. Сидять вони, підібгавши ноги, що пов'язано з м'язової гіпертонією.
Інші симптоми можуть включати:
• ненормальна поведінка (гіперактивні з безпредметними рухами, ритмічні гойдання та атетоз)
• розвиток затримки мовлення
• напади (25%) та електроенцефалографічні аномалії (50%)
• характерний запах тіла фенілоцтової кислоти, який характеризувався як затхлий або сміттєвий
• легка пігментація тіла (обумовлена відсутністю меланіну, зміна кольору волосся, шкіри та очей)
• блювання
• себорейний або екзематичний висип (незначний і зникає, коли дитина стає старшою)
• більшість дітей є гіпертонічними з гіперактивними глибокими сухожилковими рефлексами
• мікроцефалія
• видно верхню частину широко розставлених зубів
• гіпоплазія емалі
• затримка зростання
Діагностика
Діагностувати фенілкетонурію необхідно відразу після народження дитини, щоб запобігти важких наслідків захворювання. Саме тому на 4-й-5-й день життя доношеної новонародженого і на 7-й день - у недоношеного малюка у пологовому будинку береться кров для обстеження на це захворювання. Первинним діагностичним тестом для ФКУ є вимірювання рівнів фенілаланіну у краплі крові, взятої з п'яти новонародженої ноги дитини (тест Гатьрі) (рис.2).
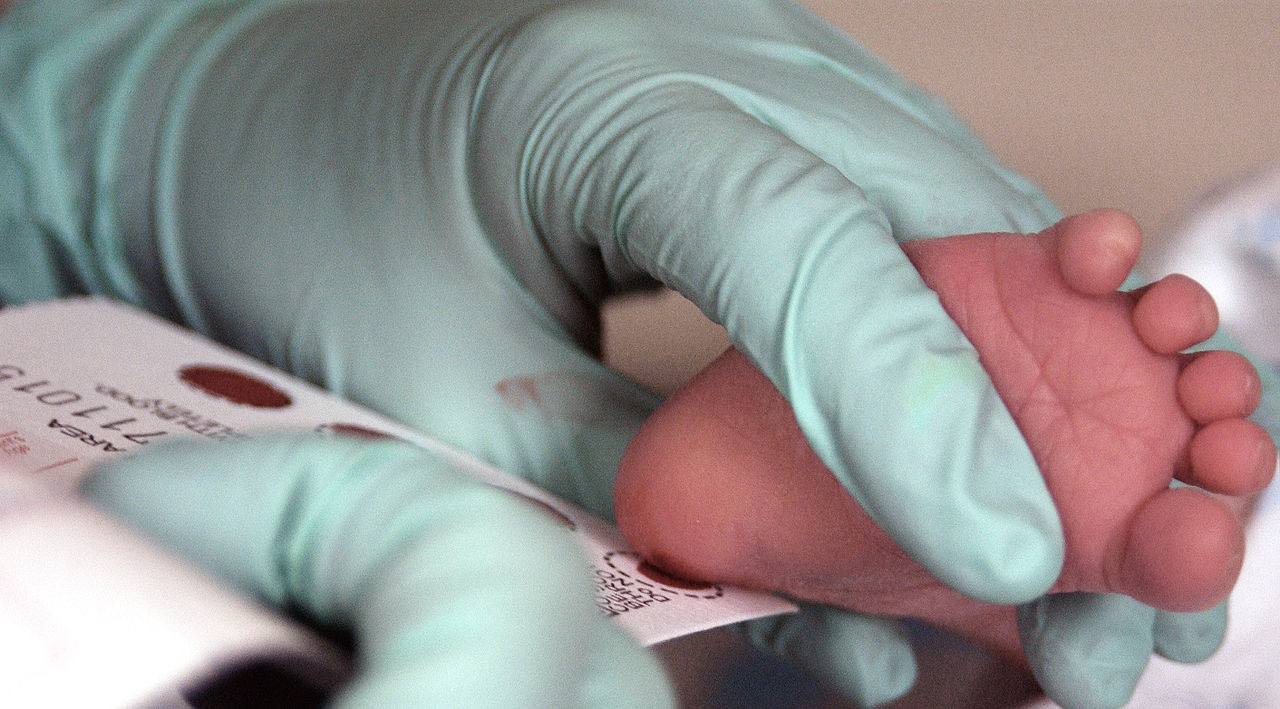
Рис. 2. - Аналіз крові немовляти на фенілкетонурію.
Через годину після годування спеціальний паперовий бланк просочується капілярною кров'ю. У тому випадку, якщо у зразку крові концентрація фенілаланіну перевищує 2,2 мг%, таку дитину направляють на дообстеження, огляд та уточнення діагнозу в медико-генетичний центр. У цьому тесті, ФКУ підтверджується появою бактерій, що ростуть навколо високих концентрацій фенілаланіну в точці крові. Існують наступні способи діагностики фенілкетонурії: виявлення у сечі малюка фенілкетонів ще в пологовому будинку або ж визначити їх у крові і сечі дорослих людей при відповідній клінічній симптоматиці. Для цього проводиться тест на толерантність до фенілаланину: людина випиває 10 гр. розчину фенілаланіну, а через деякий час визначають кількість утвореного в крові тирозину. Якщо показники становлять 9-10 грам, така людина носієм фенілкетонурії не є. Пацієнти ФКУ демонструють високий рівень фенілаланіну та низький рівень тирозину в крові.
Лікування
Лікування ФКУ рекомендується дітям, у яких діагностують рівень фенілаланіну в крові 7-10 мг / дл або більше протягом декількох днів підряд.
Дієтична терапія є найпоширенішою формою лікування пацієнтів з ФКУ. Фенілаланін фактично є незамінною амінокислотою. Дієти забезпечують дуже невелику кількість фенілаланіну та більшу кількість інших амінокислот, включаючи тирозин. Кількість допустимого фенілаланіну може трохи підвищуватися, коли дитина стає старшою. Дієти з ФКУ включають всі поживні речовини, які зазвичай потрібні для гарного здоров'я та нормального зростання (вуглеводи, жири, вітаміни та мінерали). Забороняються продукти з високим вмістом білка, такі як м'ясо, риба, курка, яйця, горіхи, боби, молоко та інші молочні продукти. Допускається невелика кількість білкової їжі (зерно та картопля) та продуктів з низьким вмістом білка (деякі фрукти та овочі, дієтичний хліб та макаронні вироби). Виключають цукор, на заміну використовують дієтична соду, яка містить штучний підсолоджувач аспартам.
Аналізи крові слід проводити рано вранці, коли рівень фенілаланіну є найвищим. Всі пацієнти з ФКУ повинні дотримуватися суворої контрольованої дієти на все життя.
Медикаментозна терапія - куван (сапроптерин дигідрохлорид), перший препарат, що допомагає регулювати ФКУ. Препарат допомагає знизити рівень фенілаланіну в крові у осіб з ФКУ, збільшуючи активність ферменту фенілаланін-4-гідроксилази. Куван ефективний лише для людей, які мають деяку активність фенілаланін-4-гідроксилази. Особи, які приймають цей препарат, повинні продовжувати дотримуватися дієти, обмеженої фенілаланіном, і проводити аналізи крові для вимірювання рівня фенілаланіну.
Пацієнти з ФКУ, які отримують ранню і послідовну дієтичну терапію, можуть розвинути досить нормальну психічну здатність в межах приблизно п'яти точок IQ їх здорових однолітків. Для порівняння, пацієнти без лікування ФКУ, як правило, мають показники IQ нижче 50.
Прогноз
ФКУ невиліковне захворювання, але рання діагностика новонароджених, ретельний контроль та тривалий строк харчування можуть запобігти розвитку серйозної психічної нездатності та допомогти пацієнтам ФКУ жити нормальним, здоровим та тривалим життям.
Профілактика
Батьки можуть провести генетичну експертизу, щоб обчислити ймовірність народження дитини з даною патологією.
Для діагностики захворювання на доклінічному рівні використовується програма масового обстеження новонароджених дітей в пологових будинках. Вона заснована на виявленні в крові збільшеного рівня фенілаланіну.
Дуже важлива рання діагностика фенілкетонурії, так як лікування захворювання з самого народження повністю купірує, і малюк виросте здоровим. Без відповідного лікування хвороба посилюється з віком, постійно накопичуються токсичні речовини з організму ж вони виводяться у мінімальній кількості.
Гомоцистинурія
Гомоцистинурія описана в 1962 р. Карсеном і Нейлом. Термін "гомоцистинурія" насправді є описом біохімічної аномалії, хоча багато з них називають гомоцистинурію хворобою. Гомоцистинурія відноситься до підвищених рівнів гомоцистеїну у сечі. Це може бути викликано різними біохімічними аномаліями і є вісім різних змін генів, які, як відомо, зумовлюють виділення з надто великим вмістом гомоцистеїну у сечі.
Етіопатогенез.
Гомоцистинурія обумовлена аутосомно-рецесивним типом успадкування. На сьогоднішній день відомо 4 типи метаболічних порушень, котрі можуть бути у основі патології, у зв`язку з чим виділяють наступні біохімічні варіанти захворювання:
-
Гомоцистинурія І – обумовлена відсутністю або зниженням активності ферменту цистионін-бета-синтази (класична гомоцистинурія).
-
Гомоцистинурія ІІ – обумовлена відсутністю або зниженням активності ферменту N5, N10-метилентетрагідрофолатредуктази.
-
Гомоцистинурія ІІІ – обумовлена низькою активністю ферменту N5-метилентетрагідрофолата.
-
Гомоцистинурія ІV – обумовлена відсутністю або зниженням активності ферменту гомоцистеїнтрансметилази, викликаних дефектом синтезу метилкоболаміну.
Найвідомішою і найпоширенішою причиною гомоцистинурії є відсутність цистатіонін-б-синтази ("класична гомоцистинурія", викликана дефіцитом цистатіонін-синтетази).
Безпосередні патогенетичні механізми патології пов'язані з порушенням метаболізму незамінної амінокислоти метіоніну. Метаболічні процеси контролюються ферментами, при інактивації яких відбувається ензиматичний блок: в крові і тканинах накопичується проміжний продукт обміну метіоніну - гомоцистину, який виводиться з сечею; при цьому також зменшується вміст цистатіонін і цистину. Можливою причиною порушення метаболічного шляху також може служити гіповітаміноз В6 і В12, а також фолієвої кислоти.
Гомоцистеїн бере участь у катаболізмі метіоніну. Метіонін є незамінною амінокислотою. Метіонін походить з дієтичного білка. Як правило, кількість споживаного метіоніну більше, ніж потреба організму. Надмірний метіонін перетворюється на гомоцистеїн, який потім метаболізується в цистатіонін; Цистатіонін потім перетворюється в цистеїн. Цистеїн виділяється з сечею. Перетворення гомоцистеїну в цистатіонін b-синтази вимагає вітаміну B6 (піридоксин). Якщо цистатіонін b-синтази відсутній, то гомоцистеїн не може бути розбитий на цистатіон і цистеїн, а замість цього накопичується гомоцистеїн, підвищений рівень гомоцистеїну та метіоніну можна знайти в крові. Підвищений рівень гомоцистеїну призводить до розвитку захворювання, якщо його не лікувати, то має негативний вплив на множинні системи, включаючи центральну нервову систему, очі, скелет і судинну систему.
Класична гомоцистинурія має аутосомно-рецесивний тип успадкування. Всесвітня частота інфікування осіб на дефіцит цистатіонін-б-синтази становить 1 на 350000.
Клінічні ознаки
Прояви гомоцистинурії наростають поступово. Діти народжуються без будь-яких специфічних відхилень. Протягом першого року життя розвивається помірно виражена гіпотрофія. Спроби усунути відставання у вазі і зростанні за рахунок додаткового введення в раціон білка у вигляді кефіру або сиру лише посилюють перебіг захворювання: наростає дефіцит маси тіла, порушується сон, дитина стає дратівливою і плаксивою, відзначається пізніше закриття тім`ячок, деформації кінцівок, затримка психомоторного розвитку.
Зазвичай яскраво виражена клініка гомоцистинурії розвивається протягом перших 10 років життя, проте часто діагноз стає очевидним вже в ранньому дитячому віці.
Порушення опорно-рухового апарату:
-
особи високі та худі з витонченням та подовженням кісток,
– довге, вузьке обличчя,
-
високе арочне піднебіння (готичне піднебіння),
-
килевидна деформація грудної клітки,
-
викривлення гомілок,
-
пальці довгі і худі (арахнодактилія),
-
кифосколіоз,
-
pectus excavatum або pectus carinatum,
-
остеопороз,
-
жорсткі суглоби,
-
плоскостопість.
Очні симптоми:
-
короткозорість,
-
підвивих кришталика,
-
тремтіння райдужки (іридодонез),
-
астигматизм,
-
глаукома,
-
катаракта,
-
відшарування сітківки,
-
атрофія зорових нервів,
-
розумова відсталість,
-
порушення м`язевого тонус,
-
гіперкінези,
-
судомний синдром,
-
порушення поведінки,
-
психічні проблеми (депресія, хронічні проблеми поведінки, хронічні обсесивно-компульсивні розлади та порушення особистості),
-
майже 50% хворих мають артеріальні тромбози (оклюзії церебральних, коронарних, ниркових і периферичних судин, а також венозні тромбози).
Найбільш частою причиною смерті, спричиненої дефіцитом цистатіонін-б-синтази, є згустки крові, що утворюються у венах і артеріях (тромбоемболії), емболії легенів та інсульти. Ускладненням дефіциту цистатіонін-б-синтази є важкий передчасний атеросклероз.
Діагностика
Дефіцит цистатіонін-б-синтази діагностується новорожденним скринінгом: підвищений рівень метіоніну в крові. Скринінг проводять шляхом збору крові із шпильки на п'ятці дитини до виписки з пологового будинку, але принаймні через 24 години після народження. Якщо рівні підвищені, то виконується наступне тестування для перевірки діагнозу. Якщо не визначено при скринінгу новонароджених, діагностика проводиться шляхом виявлення низьких рівнів цистеїну в крові та сечі.
Пацієнти з підозрою на гомоцистинурія повинні направлятися до медичного генетика для аналізу генеалогічних даних і проведення молекулярно-генетичної діагностики. Діагноз встановлюється за допомогою біохімічного дослідження крові та сечі: при гомоцистинурії у сечі, плазмі крові, лікворі виявляється значна кількість гомоцистина, підвищення вмісту метіоніну при зниженому рівні цистину. В біоптатах шкіри і печінки виявляється специфічний ферментативний дефект.
Рентгенологічне дослідження трубчастих кісток і хребта виявляє системний остеопороз. За допомогою електроенцефалографії реєструються порушення біоелектричної активності головного мозку, іноді пароксизмального характеру. Консультація офтальмолога дозволяє підтвердити характерні для гомоцистинурії порушення з боку зорової системи. Також хворі діти потребують спостереження й оцінки розвитку з боку педіатра, невролога, ортопеда, психіатра. Диференціальна діагностика гомоцистинурії здійснюється з синдромом Марфана, наслідками родової травми і внутрішньоутробними інфекціями, іншими ензимопатіями.
Муковісцидоз (кістозний фіброз)
Муковісцидоз (кістозний фіброз) - системне спадкове захворювання, обумовлене мутацією гена трансмембранного регулятора муковісцидозу і характеризується ураженням залоз зовнішньої секреції, важкими порушеннями функцій органів дихання і шлунково-кишкового тракту. Муковісцидоз успадковується за аутосомно-рецесивним типом з високою частотою популяції (від 1:1000 до 1:3500 серед новонароджених). Частота передавачів гетерозигот становить 5% і вище. Ген муковісцидозу знаходиться в 7-й хромосомі і називається CFTR. Існує понад 500 відомих змін у гені CFTR, які можуть викликати кістозний фіброз. Однак 70% всіх людей з ненормальним геном CFTR мають один і той же дефект, відомий як дельта-F508. Білок CFTR допомагає виробляти слиз. Мюкус - це складна суміш солей, води, цукрів та білків, що очищає, змащує та виконує захисну фукцію у організмі (легенях та підшлунковій залозі). Муковісцидоз впливає на здатність організму транспортувати сіль і воду до клітини і виходити з неї.
Кістозний фіброз головним чином вражає людей білого північного європейського походження.
Якщо обоє батьків гетерозиготні і носії мутованого гена, то ризик народження хворої на муковісцидоз дитини становить 25%. За даними досліджень частота гетерозиготного носійства патологічного гена дорівнює 2-5%.
Патогенез
Ідентифіковано близько 1000 мутацій гена муковісцидозу. Наслідком мутації гена є порушення структури та функції білка, що отримав назву трансмембранний регулятор муковісцидозу (ТРМ). Наслідком цього є згущення секретів залоз зовнішньої секреції, утруднення евакуації секрету і зміна його фізико-хімічних властивостей, що, в свою чергу, і обумовлює клінічну картину захворювання. Зміни в підшлунковій залозі, органах дихання, шлунково-кишковому тракті реєструються вже у внутрішньоутробному періоді і з віком пацієнта неухильно наростають. Виділення в'язкого секрету екзокринними залозами призводить до утруднення відтоку і застою з подальшим розширенням вивідних проток залоз, атрофією залозистої тканини і розвитком прогресуючого фіброзу. Активність ферментів кишечника і підшлункової залози значно знижена. Поряд з формуванням склерозу в органах має місце порушення функцій фібробластів. Встановлено, що фібробласти хворих на муковісцидоз продукують циліарний фактор, або М-фактор, який володіє антіціліарною активністю - він порушує роботу війок епітелію.
В даний час розглядається можлива участь у розвитку патології легень у разі муковісцидозу генів, відповідальних за формування імунної відповіді (зокрема, генів інтерлейкіну-4 (IL-4) і його рецепторів), а також генів, що кодують синтез оксиду азоту (NO) в організмі .
Клінічні ознаки
Найбільш серйозні наслідки муковісцидозу виявляються у шлунково-кишковій системі, дихальних шляхах, потових залозах та чоловічії родукції. Симптоми розвиваються поступово.
Основними формами захворювання є:
- Переважно легенева форма (респіраторна, бронхолегенева);
- Переважно кишкова форма;
- Змішана форма з одночасним ураженням шлунково-кишкового тракту і органів дихання;
- Меконієва непрохідність кишечника;
- Атипові і стерті форми (набряково-анемічна, циротична та ін.)
Респіраторна форма.
Першими симптомами бронхолегеневої форми муковісцидозу є млявість, блідість шкірних покривів, недостатня прибавка маси тіла при задовільному апетиті. У деяких випадках (тяжкий перебіг) з перших днів життя у хворого з'являється покашлювання, яке поступово посилюється і набуває кашлюкоподібного характеру. Кашель супроводжується виділенням густого харкотиння, яке при нашаруванні бактеріальної флори стає згодом слизово-гнійним.
Підвищена в'язкість бронхіального секрету призводить до розвитку мукостаза і закупорці дрібних бронхів і бронхіол, що сприяє розвитку емфіземи, а при повній закупорці бронхів - формуванню ателектазів. У дітей раннього віку до патологічного процесу швидко втягується паренхіма легені, що призводить до розвитку важкої, затяжної пневмонії зі схильністю до абсцедування. Ураження легень завжди двостороннє.
При об'єктивному обстеженні відзначаються вологі дрібно-та средньопухирчасті хрипи, а при перкусії вислуховується коробковий відтінок звуку. У хворих може розвинутися токсикоз і навіть клініка шоку на фоні захворювань, що протікають з високою температурою тіла, або у спекотну пору року при значній втраті натрію і хлору з потом. Надалі пневмонія набуває хронічного перебігу, формуються пневмосклероз і бронхоектази, з'являються симптоми «легеневого серця», легенева і серцева недостатність.
У клінічній картині звертає на себе увагу зовнішній вигляд хворого: бліда шкіра з землистим відтінком, акроціаноз, загальний ціаноз, задишка у спокої, бочкоподібна форма грудної клітки, деформації грудини по типу «клиноподібної» і деформації кінцевих фаланг пальців за типом «барабанних паличок» (рис.3), обмеження рухової активності, зниження апетиту та зменшення маси тіла.
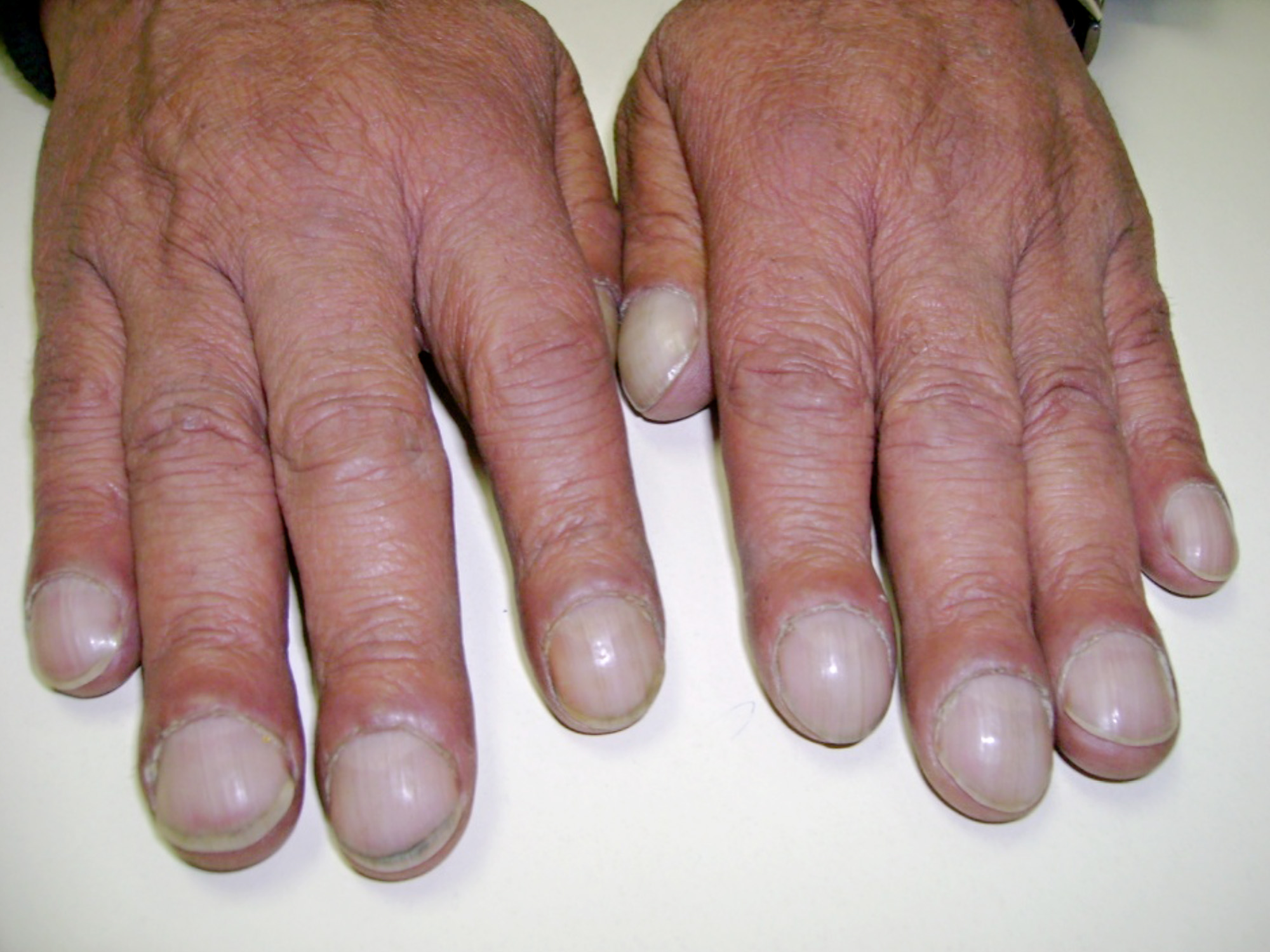
Рис. 3. - Симптом барабанних паличок і часових скелець при муковісцидозі.
Рідкими ускладненнями при муковісцидозі є пневмо-і піопневмоторакс, легенева кровотеча. При більш сприятливому перебігу муковісцидозу, що спостерігається у разі маніфестації захворювання у більш старшому віці, бронхолегенева патологія проявляється повільно прогресуючим деформуючим бронхітом з помірно вираженим пневмосклерозом.
У разі тривалого перебігу захворювання до патологічного процесу залучається носоглотка: синусит, аденоїдні вегетації, поліпи носа, хронічний тонзиліт. Рентгенологічне дослідження легенів при муковісцидозі дозволяє виявити поширені перибронхіальні, інфільтративні, склеротичні зміни і ателектази на тлі вираженої емфіземи. При бронхографії відзначається наявність каплеподібних бронхоектазів, відхилень бронхів і зменшення числа дрібних розгалужень, бронхи 3-6-го порядку мають вигляд чоток. При бронхоскопії нерідко виявляють невелику кількість густого в'язкого харкотиння, що розташовується у вигляді ниток у просвітах великих бронхів. Мікробіологічне дослідження харкотиння у хворих на муковісцидоз дозволяє виділити золотистий стафілокок, гемофільну та синьогнійну палички. Наявність синьогнійної палички у мокроті є прогностично несприятливою ознакою для пацієнта.
Кишкова форма
Клінічна симптоматика кишкової форми обумовлена секреторною недостатністю шлунково-кишкового тракту. Порушення ферментативної активності шлунково-кишкового тракту особливо яскраво виражене після переведення дитини на штучне вигодовування або прикорм і виявляється недостатнім розщепленням і всмоктуванням білків, жирів і меншою мірою вуглеводів. У кишечнику переважають гнильні процеси, що супроводжуються накопиченням газів, які призводить до здуття живота, частих дефекацій, поліфекалія (добовий обсяг калових мас у 2-8 разів може перевищувати вікову норму). Після того, як хвору на муковісцидоз дитини починають висаджувати на горщик, нерідко відзначається випадання прямої кишки (у 10-20% хворих). Хворі скаржаться на сухість у роті, що обумовлено високою в'язкістю слини. Хворі з важкістю пережовують суху їжу, а під час їжі вживають значну кількість рідини. Апетит у перші місяці збережений або навіть підвищений, але внаслідок порушення процесів травлення у хворих швидко розвивається гіпотрофія, полігіповітаміноз. М'язовий тонус і тургор тканин знижений. Хворі скаржаться на біль у животі різного характеру: переймоподібний - при метеоризмі, м'язовий - після нападу кашлю, біль у правому підребір'ї - при наявності правошлуночкової недостатності, біль в ділянці епігастрію обумовлений недостатньою нейтралізацією шлункового соку у дванадцятипалій кишці при зниженій секреції підшлунковою залозою бікарбонатів.
Порушення нейтралізації шлункового соку може стати причиною розвитку виразкової хвороби дванадцятипалої кишки або виразкового процесу у тонкому кишечнику. Ускладненнями кишкової форми муковісцидозу можуть бути вторинна дисахаридазна недостатність, кишкова непрохідність, вторинний пієлонефрит і сечокам'яна хвороба на тлі обмінних порушень, латентно протікає цукровий діабет при ураженні інсулярного апарату підшлункової залози. Порушення білкового обміну призводить до гіпопротеїнемії, що стає причиною розвитку у деяких випадках у дітей грудного віку набрякового синдрому.
Гепатомегалія (збільшення печінки) обумовлена холестазом. При біліарному цирозі у клінічній картині можна спостерігати жовтяницю, свербіж шкіри, ознаки портальної гіпертензії, асцит. Цироз печінки у деяких хворих може розвинутися і без холестазу.
Змішана форма
Змішана форма муковісцидозу є найбільш важкою і включає клінічні симптоми як легеневої, так і кишкової форм. Зазвичай з перших тижнів життя хворого відзначаються важкі повторні бронхіти і пневмонії із затяжним перебігом, постійний кашель, кишковий синдром і різкі розлади харчування. Клінічна картина муковісцидозу відрізняється значним поліморфізмом, що і визначає варіанти перебігу захворювання. Відзначено залежність тяжкості перебігу муковісцидозу від строків появи перших симптомів - чим молодша дитина до моменту маніфестації хвороби, тим важче його перебіг і більш несприятливий прогноз. Враховуючи поліморфізм клінічних проявів муковісцидозу, тяжкість перебігу прийнято оцінювати у більшості випадків характером і ступенем ураження бронхолегеневої системи.
Меконієва непрохідність.
У 30-40% хворих муковісцидоз діагностовано в перші дні життя у вигляді меконіевої непрохідності. Дана форма захворювання обумовлена відсутністю трипсину, що призводить до скупчення у петлях тонкого кишечника (найчастіше в ілеоцекальній ділянці) щільного, в'язкого по консистенції меконію.
У здорового новонародженого первородний кал відходить на першу, рідше - другу добу після народження. У хворої дитини відсутнє виділення меконію. До другого дня життя дитина стає неспокійною, живіт роздутий, відзначаються зригування і блювання з домішкою жовчі. Через 1-2 дні стан новонародженого погіршується: шкірні покриви стають сухі і бліді, на шкірі живота з'являється виражений судинний малюнок, тургор тканин знижений, занепокоєння змінюється млявістю і адинамією, наростають симптоми інтоксикації і ексикозу.
При об'єктивному обстеженні пацієнта відзначаються задишка і тахікардія, при перкусії живота - тимпаніт, при аускультації перистальтика не прослуховується. Оглядова рентгенограма органів черевної порожнини дозволяє виявити роздуті петлі тонкого кишечника і спалі відділи у нижній частині живота.
Ускладненням меконіевої непрохідності може бути перфорація кишечника з розвитком меконіевого перитоніту. Нерідко на тлі непрохідності кишечника у хворих на муковісцидоз на 3-4-у добу життя приєднується пневмонія, яка має затяжний характер. Кишкова непрохідність може розвинутися і в більш пізньому віці хворого.
Потові залози
Потовиділення рясніше ніж зазвичай, і вимірювання солоності пота людини є найважливішим діагностичним тестом для муковісцидозу. Більш старші діти та дорослі з муковісцидозом компенсують цю додаткову втрату солі, вживаючи більше солоної їжі. Немовлята і маленькі діти страждають від наслідків (гаряча прострація). Теплопростація відмічена летаргією, слабкістю, втратою апетиту і повинна розглядатися як надзвичайний стан.
Народжуваність
98% чоловіків з кістозним фіброзом є стерильними, через повну непрохідність або відсутність трубки, яка несе сперму з яєчок. Більшість жінок з кістозним фіброзом є родючими. У хлопчиків та дівчат, статеве дозрівання затримується через наслідки поганого харчування або хронічної легеневої інфекції.
Діагностика
Діагноз муковісцидозу визначається даними клінічних та лабораторних методів обстеження пацієнта. З метою ранньої діагностики муковісцидоз входить до програми обстеження новонароджених на спадкові і вроджені захворювання. Досліджують рівень імунореактивного трипсину у сухій плямі крові. При позитивному результаті тест повторюють на 21-28 день життя. У разі повторного позитивного результата призначають потовий тест.
Для постановки діагнозу захворювання необхідна наявність чотирьох основних критеріїв:
хронічний бронхолегеневий процесс;
кишковий синдром;
випадки муковісцидозу у сибсів;
позитивні результати потового тесту.
Піт для дослідження збирають після електрофорезу з пілокарпіном. Мінімальна кількість поту, необхідна для отримання достовірного результату, становить 100 мг. Різниця між показниками натрію і хлору у пробі не повинна перевищувати 20 ммоль/л, у іншому випадку дослідження повторюють. При відпрацьованій методиці допустимо визначення одного з іонів. У здорових дітей концентрація іонів натрію і хлору у поті не повинна перевищувати 40 ммоль/л. Діагностично достовірним критерієм муковісцидозу є вміст іонів хлору вище 60ммоль/л і натрію - вище 70 ммоль/л. Для підтвердження діагнозу потрібен позитивний триразовий потовий тест з вмістом хлоридів поту вище 60 ммоль/л. Важливе значення у діагностиці муковісцидозу має копрологічне дослідження.
У копрограмі хворого на муковісцидоз найбільш характерною ознакою є підвищений вміст нейтрального жиру, але можлива наявність м'язових волокон, клітковини та крохмальних зерен, що дозволяє визначити ступінь порушення ферментативної активності залоз шлунково-кишкового тракту. Під контролем даних копрологічного дослідження проводять корекцію дози панкреатичних ферментів.
Орієнтовними методами для діагностики муковісцидозу є визначення протеолітичної активності калу рентгенологічним тестом, активності ферментів підшлункової залози у дуоденальному вмісті, концентрації натрію у нігтях і секреті слинних залоз. В якості скринінг-тесту у періоді новонародженості використовують метод визначення підвищеного вмісту альбуміну у меконію - меконіальний тест (у нормі вміст альбуміну не перевищує 20 мг на 1 г сухої маси).
Особливе місце у діагностиці займає молекулярно-генетичне тестування. В даний час за наявністю відомих мутацій у гені CFTR доступні ідентифікації 65-75% хворих на муковісцидоз, що не дає можливості використовувати для верифікації діагнозу захворювання тільки молекулярно-генетичне обстеження.
Загальні ознаки: відставання у фізичному розвитку, рецидивуючі хронічні захворювання органів дихання, поліпи носа, хронічний гайморит, хронічний бронхіт, рецидивуючий панкреатит, дихальна недостатність, хронічні коліти, холецистити у родичів.
Потовий тест: іонофорез з пілокарпіну.
Підвищення хлоридів більше 60 ммоль/л - ймовірний діагноз;
концентрація хлоридів > 100 ммоль/л - достовірний діагноз.
При цьому різниця у концентрації хлору і натрію не повинна перевищувати 8-10 ммоль/л. Потовий тест для постановки остаточного діагнозу повинен бути позитивним не менше трьох разів. Потову пробу необхідно проводити кожній дитині з хронічним кашлем.
Хімотрипсин у стільці: проба не стандартизована - нормативні значення розробляються у конкретній лабораторії.
Визначення жирних кислот у калі: в нормі менше 20 ммоль/день. Межові значення - 20-25 ммоль/день. Проба позитивна при зниженні функції підшлункової залози не менш ніж на 75%.
ДНК-діагностика найбільш чутлива і специфічна. Помилкові результати отримують у 0,5-3% випадків.
Пренатальна ДНК-діагностика: дослідження ізоензимів тонкокишечної лужної фосфатази з навколоплідних вод, можливо з 18-20 тижня вагітності. Хибнопозитивні і хибнонегативні значення отримують у 4% випадків.
Лікування муковісцидозу (кістозного фіброзу)
Лікування муковісцидозу симптоматичне. Важливе значення має харчування хворого. Добовий калораж повинен на 10-30% перевищувати вікову норму за рахунок збільшення у раціоні білкового компонента. Потребу у білках задовольняють вживанням у їжу м'яса, риби, яєць, сиру. Споживання жирів значно обмежують. Можна використовувати жири, до складу яких входять жирні кислоти з середнім розміром ланцюга, так як їх засвоєння не залежить від активності ліпази підшлункової залози.
При дефіциті дисахаридаз у тонкому кишечнику з раціону виключають відповідні цукри (найчастіше лактозу). Їжу хворим завжди підсолюють, особливо у спекотну пору року і у разі високої температури, враховуючи великі втрати солей з потом. Хворому забезпечують вживання достатньої кількості рідини. У харчування повинні бути включені продукти, які містять багато вітамінів, фруктові та овочеві соки, вершкове масло.
У обов'язковому порядку здійснюють корекцію порушеної функції підшлункової залози шляхом застосування панкреатину або комбінованих препаратів, що містять поряд з панкреатином інші кишкові ферменти і ліпотропні речовини (полізім, панзинорм, мексаза та ін.) Дозу ферментних препаратів підбирають індивідуально, орієнтуючись на дані копрологічного дослідження.
Показниками оптимального підбору дози служать нормалізація стільця і зникнення у калі нейтрального жиру. Початкова доза препарату становить 2-3 г на добу. Дозу поступово підвищують до появи позитивного ефекту. Для розрідження секретів шлунково-кишкового тракту і поліпшення їх відтоку застосовують ацетилцистеїн у таблетках і гранулах, що показано при холестазі, в'язкому дуоденальному вмісті, неможливості провести зондування. Лікування легеневого синдрому включає комплекс заходів, спрямованих на розрідження мокроти і видалення її з бронхів. З цією метою застосовують фізичні, хімічні та інструментальні методи. Муколітична терапія проводиться щодня протягом всього життя пацієнта. Ефективність лікування підвищується при паралельному використанні аерозольних інгаляцій, ЛФК, вібраційного масажу, постурального дренажу. Кількість і тривалість інгаляцій визначають тяжкістю стану хворого. Як муколітичних препаратів можна використовувати соляно-лужні суміші (1-2% сольовий розчин - хлорид і карбонат натрію), бронхолітичні препарати, ацетилцистеїн (на одну інгаляцію 2-3 мл 7-10% розчину), пульмозім (дорназа альфа). Постуральний дренаж проводиться щоранку, вібраційний масаж - не менше 3 разів на добу.
Лікувальна бронхоскопія з промиванням бронхів ацетилцистеїном і ізотонічним розчином хлориду натрію показана як екстрена процедура у разі відсутності ефекту вищеописаної терапії. У періоди загострення захворювання, при наявності гострої пневмонії або гострої респіраторної вірусної інфекції показано застосування антибактеріальної терапії.
Антибактеріальні засоби вводять парентерально (напівсинтетичні пеніциліни, цефалоспорини другого і третього покоління, аміноглікозиди, хінолони) і у вигляді аерозолів (аміноглікозиди: гентаміцин, тобраміцин). Враховуючи схильність пневмоній при муковісцидозі до затяжного перебігу, курс антибіотиків має становити не менше одного місяця, а іноді й більше.
При важкому перебігу пневмонії застосовують кортикостероїдні препарати протягом 15 діб - 2 місяців. Преднізолон призначають із розрахунку 10-15 мг /кг на добу протягом 10 - 15 днів. Потім дозу поступово знижують.
Антибіотики застосовують протягом всього курсу кортикостероїдної терапії. Поряд з антибактеріальною та муколітичною терапією проводять повний комплекс лікувальних заходів, спрямованих на боротьбу з гіпоксією, серцево-судинними порушеннями, змінами кислотно-основного стану. При організації диспансерного спостереження за хворими на муковісцидоз в амбулаторних умовах необхідно здійснювати контроль за стільцем і масою тіла хворого, регулярно (1 раз на 3 місяці) проводити копрологічне дослідження з метою корекції дози препаратів підшлункової залози, навесні і при загостренні процесу призначати курси вітамінотерапії (виправдане призначення подвійної дози жиророзчинних вітамінів А, Е, D у вигляді водних розчинів).
Родичів хворого необхідно навчити прийомам постурального дренажу, вібраційного масажу та догляду за пацієнтом. Поряд із заняттями лікувальною фізкультурою рекомендовані дозовані фізичні навантаження і заняття спортом. При стійкій ремісії протягом 6 місяців дозволяється проведення профілактичних щеплень.
Профілактика
Найбільш ефективними методами профілактики є медико-генетична консультація, а також раннє виявлення хвороби у періоді новонародженості, хоча в цьому випадку мова йде не про профілактику хвороби, але про попередження її тяжких ускладнень.
Вроджений синдром гіпотиреозу
Вроджений гіпотиреоз — захворювання, викликане зниженням або повним випадінням функції щитовидної залози, яке існує при народженні.
Вроджений гіпотиреоз являє собою особливу форму гіпофункції щитовидної залози. Вона вражає пацієнтів у той період розвитку, коли вся нервова система є дуже чутливою і може пошкодитися через тривалу нестачу гормонів щитовидної залози. Якщо безпосередньо після народження не виявити порушення і не скоригувати його, то це приведе до розвитку кретинізму.
Причини
У більшості випадків спричиняється зниження функції щитоподібної залози через порушення розвитку органа в ембріональному періоді (агенезія чи дисгенезія). При цьому розвивається повна функціональна неспроможність тканин щитоподібної залози або ж вона занадто мала. Причина цих пошкоджень невідома. Лише у поодиноких випадках змогли встановити генні мутації, які відігравали певну роль для розвитку щитоподібної залози, серед них FOXE1, що кодує тиреоїдний фактор транскрипції 2 (TTF-2) і PAX8-ген, що кодує Paired-Box-Protein 8 (PAX-8).
Поряд з цим бувають порушення синтезу гормонів щитоподібної залози, яка закладена у всьому іншому правильно. Вроджені порушення синтезу тироксину успадковуються за аутосомно-рецесивним типом, дефекти рецепторів до гормонів щитоподібної залози успадковуються аутосомно-домінантно. У загальному близько 85% всіх вроджених гіпофункцій щитоподібної залози є спорадичними, інші 15% — спадково обумовленими.
В окремих випадках гіпофункція має перехідну природу. Такі випадки спричиняються попаданням через плаценту до дитини медикаментів чи материнських блокуючих антитіл, дифицітом йоду чи навпаки надмірним його споживанням. Порушення гіпоталамо-гіпофізарної системи — так звана центральна, вторинна чи третинна гіпофункція — зі зниженою продукцією тиреотропного гормону (ТТГ) чи тиреотропін-рилізінг-гормону (ТРГ, тиреоліберин) є екстремально рідкісним явищем, однак вони не виявляються при звичайному скринінговому обстеженні.
Клінічні ознаки
Гормони щитоподібної залози мають дуже велике значення для розвитку майже всіх систем органів і більшість дітей при народжені спочатку нічим не відрізняються. Це обумовлено материнськими гормонами щитоподібної залози, які проникають через плаценту, і як мінімум частково захищають навіть ті плоди, в яких щитовидна залоза повністю відсутня. У пуповинній крові цих дітей концентрація тироксину (Т4) становить від 33% до 50% від рівня здорових дітей. У 5% найважчих випадків можна виявити велике тім'ячко, значне незрощення черепних швів, великий язик і пупкову грижу. З посиленням розпаду материнського тироксину зявляються нові симптоми. Такі немовлята погано смокчуть, страждають закрепами, байдужі до навколишнього середовища і багато сплять. Часто їх потрібно спеціально будити для годування. Голос може бути грубим і сиплим, шкіра на дотик холодна, слабкий тонус м'язів (гіпотонія), рефлекси ослаблені. Жовтяниця новонароджених, що обумовлена сповільненим дозріванням печінки, може затягнутися. Збільшену щитовидну залозу (зоб, струма) знаходять у 5-10% дітей, найчастіше при вродженому порушенні синтезу тироксину (рис.4).
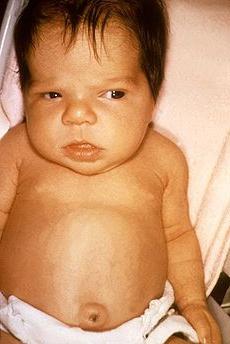
Рис. 4 – Зовнішній вигляд дитини з вродженим синдромом гіпотиреозу
Якщо захворювання не розпізнається і не лікується, на другому-третьому місяці життя проявляється затримка росту. Чим пізніше розпочате лікування, тим нижчим буде розумовий розвиток дитини. Якщо почати лікування у перші три місяці, в середньому коефіцієнт інтелекту (IQ) становитиме 89 (розмах 64-107), якщо почати лікування на 4-6 місяці — 71 (розмах 35-96) і якщо лікування вперше застосувалося після шести місяців 54 (розмах 25-80). Іншими віддаленими наслідками є порушення координації дрібної і великої моторики, порушення рівноваги (атаксії), зниження і підвищення м'язового тонусу, порушення мовлення, уваги, косоокість.
Діагностика
Оскільки рання діагностика є надзвичайно важливою для нормального розвитку дітей, діагноз встановлюється у більшості розвинених країн через масове дослідження новонароджених (скринінг новонароджених). В Україні визначають вміст тиреотропного гормону (ТТГ) в так званій сухій краплі. При недостатній продукції гормонів щитовидної залози ТТГ значно підвищується. Правда, при цьому методі упускаються рідкісні випадки центральнообумовленого вторинного чи третинного гіпотиреозу.
У США більшість скринінг-програм визначають спершу концентрацію Т4 і тільки при зниженні його додатково визначають тиреотропін.
Забір крові мусить бути обов'язково проведений уже на третю добу життя. Якщо новонародженого відпускаються раніше чи переводять, можна провести забір ще раніше. Але тоді аналіз потрібно ще раз повторити.
В позитивному випадку потрібно терміново провести підтверджуючий тест на визначення тиреотропіну, вільного Т3 і Т4.
Якщо результат підтвердить гіпофункцію щитовидної залози, можна провести додаткові обстеження для з`ясування природи захворювання. До них належать, крім визначення тиреоглобуліну, обстеження тканин щитовидної залози і процесу утворення гормонів. Перед усім це візуалізаційні методи: УЗД і сцинтіграфія щитовидної залози. Оскільки результати досліджень не мають ніякого впливу на лікування, їх не потрібно проводити в обов'язковому порядку. Якщо є дані про захворювання щитовидної залози у матері, можна визначити схожість форм аутоантитіл щитовидної залози у матері і дитини. Якщо схожість підтвердиться, захворювання може носити перехідний (тимчасовий) характер. При виникненні підозри на надлишок чи недостачу йоду, як причину гіпотиреозу, можна дослідити рівень йоду у сечі.
Лікування
Враховуючи незворотність пошкодження нервової системи при вродженому гіпотиреозі, лікування потрібно починати якомога раніше. Не потрібно очікувати результатів необов'язкових методів обстежень, лікування мусить починатись безпосередньо після забору крові для підтвердження діагнозу при позитивнуму результату скринінгу. Мета лікування полягає в якнайшвидшій нормалізації вмісту Т4 з наступною нормалізацією рівня ТТГ у крові. Усунення дефіциту гормону вирівнюється призначенням левотироксину у відносно високих вихідних дозах. Через два тижні можна зменшити дозування відповідно до результатів контрольного рівня ТТГ і Т4 в крові. У подальшому доза гормону має бути адаптована відповідно до підвищенної потреби обумовленої фізичним розвитком дитини.
При виникненні сумнівів щодо стійкості гіпотиреозу після першого дня народження потрібно зробити перерву у вживанні гормону. На думку американських фахівців це доцільно зробити на четвертому році життя.
В іншому випадку замісна терапія має проводитися пожиттєво. Якщо доказано, що причиною гіпофункції є нестача йоду, лікування проводять препаратами йоду.
Прогноз
Прогноз для хворих дітей залежить від терміну встановлення діагнозу. Якщо гіпофункція виявлена протягом перших двох тижнів і адекватно пролікована, то в дорослому віці спостерігається лише невелика різниця у рівні розумового розвитку, шкільній успішності і нейрофізіологічних тестах у порівнянні зі здоровими однолітками. Якщо лікування почалося дещо пізніше, фізичний розвиток може ще нормалізуватися, однак розумовий розвиток залишиться зниженим.
Нейрофіброматоз
Нейрофіброматоз — полісистемне, поліорганне, спадкове захворювання з аутосомно-домінантним моногенним типом успадкування, яке відносять до групи факоматозів. Останні представляють групу спадкових захворювань, при яких загальним фактором є ураження нервової системи та шкіри, рідше — кісткової тканини, очей, інколи — внутрішніх органів.
Етіологія
Достеменно невідома. На сьогодні розглядають ендокринну, дизонтогенетичну і неврогенну теорії виникнення нейрофіброматозу. У розвитку нейрофіброматозної тканини беруть участь як нервові волокна, так і сполучнотканинні елементи ендо- і периневрію. Найбільше аргументів є на користь дизембріогенетичної теорії, згідно з якою доказом участі у розвитку нейрофіброматозу ектодерми є ураження нервової системи і шкіри, а мезодерми — кісткової тканини. Є теорії про те, що процес запускається внаслідок хромосомних аберацій у 22-й чи 17-й хромосомах.
Патогенез
Підґрунтям патологічних змін є порушення утворення з гена протеїну NF1 або NF2 (нейрофібромін). Цей білок є супресором пухлин і тому служить регулятором сигналу проліферації та диференціації клітин. Дисфункція нейрофіброміну може впливати на регуляцію і спричиняти неконтрольовану проліферацію клітин.
Клінічні ознаки
|
Р |
 ис.
5 – Шкірні прояви нейрофіброматозу
ис.
5 – Шкірні прояви нейрофіброматозу|
Рис. 6 – Очні симптоми нейрофіброматозу |
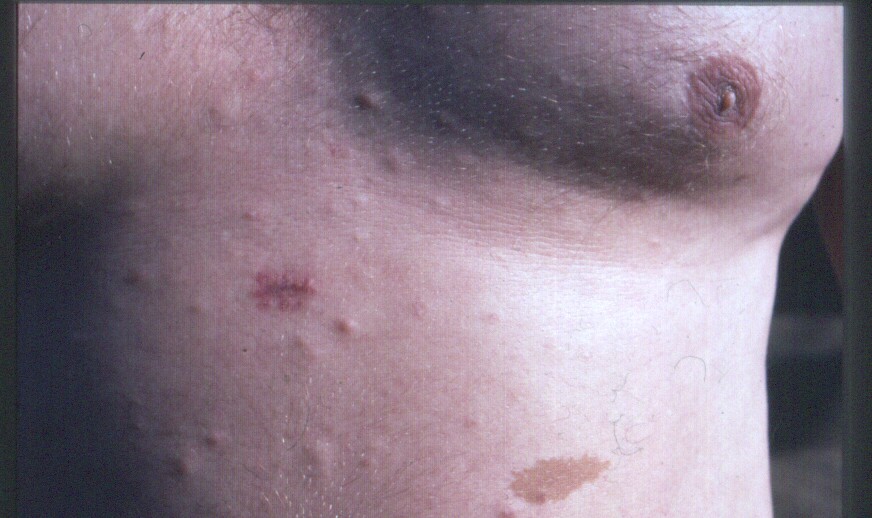
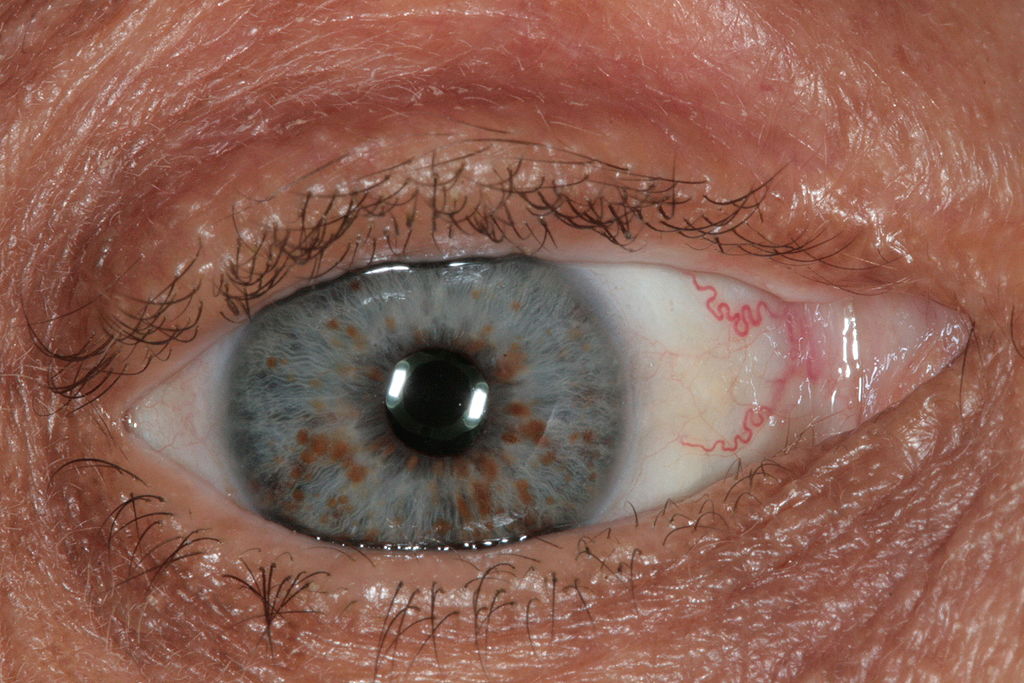
Перша ознака — пігментні плями (рис.5). Вони мають світло-кавовий колір, улюблена локалізація — внутрішня поверхня кінцівок, область спини, паху. Над поверхнею шкіри не піднімаються. Поряд з пігментними плямами можуть бути депігментовані ділянки шкіри, які нагадують вітиліго. На очах можуть з'явитися вузлики Ліша, які проявляються плямами відтінку світлої кави на райдужці ока біля зіниць (рис.6).
Другою ознакою є пухлини шкіри і підшкірної жирової клітковини, які виявляють у двох третин. Вони проявляються у трьох формах:
Слоновість. Для неї при нейрофіброматозі характерна деформація обличчя через пухлину, яка охоплює кілька ділянок. Частіше це відбувається у скроневій, привушній, щічній областях. Таку деформацію виявляють одразу після народження, вона збільшується протягом перших 2-3 років життя. Шкіра над нею спочатку звичайного забарвлення, зберігає свій тургор і еластичність, однак з віком темнішає, атрофується і звисає складками.
Вузлуваті утворення (рис.7).
Р
|
 ис.
7 – Вузліваті утворення
ис.
7 – Вузліваті утворення
Масивні пігментовані нейрофіброми. Вони частіше локалізуються на волосистій частині голови. Шкіра тут щільно пов'язана з пухлиною, пігментована — темно-коричневого забарвлення, волосся з часом у цій ділянці може випадати. Якщо пухлина локалізується на обличчі, вона також має таке забарвлення. Функція шкірних залоз підвищена, тому їхній секрет накопичується у складках пухлини, що надає обличчю масляний вигляд, ускладнює його гігієну. Для цієї групи уражень інші ознаки нейрофіброматозу не типові.
Третя ознака — пухлини нервів. При цьому уражається периферична нервова система. Пухлини являють собою вузли веретеноподібної або неправильної форми, які міцно охоплюють нервові волокна. Клінічно пухлини нервів можуть ніяк не проявлятися, лише при хірургічному втручанні можливо побачити їх у вигляді білих безкапсульних новоутворень і видалити. В окремих випадках хворих турбує парестезія, анестезія або гіперестезія навколишніх тканин.
Четвертою ознакою є фізичні й психічні порушення, зокрема пізній розвиток вторинних статевих ознак, акромегалія, порушення діяльності вищої нервової системи у вигляді псевдопсихоорганічного синдрому, що проявляється олігофренією, затримкою розвитку мови і пізнавальних функцій, якы спостерігають у більшості пацієнтів.
Усі чотири ознаки входять до діагностичної тетради Дарньє. Часто спостерігають різні аномалії зубо-щелепної системи, що проявляються деформаціями альвеолярного відростка — порушеннями прикусу, гіпоплазією емалі, адентією, флюорозом зубів, макроглосією.
Слід розуміти, що усі ознаки рідко відбуваються разом, бо різним типам нейрофіброматозу притаманні певні особливості.
На сьогодні розрізняють 4 типи нейрофіброматозу:
Нейрофіброматоз І типу
— один з типів нейрофіброматозу. Є найпоширенішою моногенною спадковою хворобою, яка призводить до виникнення пухлин у людини. NF1 є офіційним міжнародним генетичним символом нейрофіброматозу цього типу. Тип успадкування – аутосомно-домінантний. Частота виникнення захворювання у чоловіків і жінок однакова, спостерігається приблизно у кожного 3500 новонародженого. Ризик успадкування дитиною даної патології при наявності НФ1 у одного з батьків дорівнює 50%, у обох - 66,7%. Ген картирований у 17-й хромосомі.
Нейрофіброматоз II типу
— аутосомно-домінантна спадкова хвороба, один з типів нейрофіброматозу. Характеризується утворенням численних доброякісних пухлин, переважно з шваннівських клітин (шванном), рідше менінгіом і епендіом. NF2 є офіційним міжнародним генетичним символом нейрофіброматозу цього типу
Нейрофіброматоз III тип зустрічається рідко, характеризується утворенням нейрофібром на долонях, блідими, відносно великими пігментаціями відтінку кави з молоком, двобічними невриномами присінково-завиткових нервів, менінгіомами задньої черепної ямки і верхньошийного відділу хребта, спинальними та параспинальнимих нейрофібромами, відсутністю вузликів Ліша (гамартом райдужки) і пухлини ЦНС, що швидко розвиваються на другому або третьому десятку життя.
Нейрофіброматоз IV тип також є рідкісним. Клінічно нагадує нейрофіброматоз I типу, але немає вузликів Ліша.
Діагностика
Діагностика нейрофіброматозу ґрунтується на скаргах, даних клінічного (наявності ознак тетради Дарньє), лабораторного, патоморфологічного та інструментального обстеження. Наявність в сім'ї подібних випадків полегшує діагностику.
Лабораторно-інструментальне обстеження
У половини хворих в клінічному аналізі крові виявляють непостійний лейкоцитоз і еозинофілію. Для діагностики нейрофіброматозу використовують на сьогодні такі підтверджуючі методи:
МРТ
ЕЕГ
Дослідження очей за допомогою щілинної лампи
Генетичне тестування
Патогістологічні методи
Усі МРТ- і КТ-ознаки при цьому поділяють на три групи:
перша группа — загальні зміни скелета, що носять вроджений характер. До них відносять потовщення, дисплазію і подовження кісток кінцівок, черепа і лицевого скелета.
друга группа — місцеві зміни, що виникають внаслідок утворення пухлин з кісток. Проявляються вони у вигляді деструкцій, екзостозів, періостальних нашарувань, простих дефектів.
третя группа — «гіпертензивні» зміни, що виникають через підвищення внутрішньочерепного тиску в черепі при оклюзії лікворних шляхів пухлинами
Лікування
Лікування дітей з нейрофіброматозом багатоетапне, тривале, вимагає втручання різних фахівців (стоматолога, окуліста, нейрохірурга, педіатра тощо). Лікування спрямоване на відновлення порушених функцій жування, ковтання, дихання, зору, усунення деформацій м'яких тканин і кісток.
Прогноз
Прогноз змінюється в залежності від типу пухлини, яка розвивається. Оскільки пухлини ростуть, вони починають знищувати навколишні нерви та структури. Це руйнування може призвести до сліпоти, глухоти, дедалі меншої рівноваги та ускладнення координації. Деформації кісток та хребта можуть перешкоджати ходьбі та руху. Коли розвиваються ракові захворювання, прогноз погіршується залежно від конкретного типу раку.
Синдром Елерса-Данлоса
Синдром Елерса-Данлоса (недосконалий десмогенез Русакова, синдром Черногубова - Елерса - Данлоса) - це група спадкових системних захворювань сполучної тканини, викликаних дефектом у синтезі колагену. Залежно від окремої мутації, серйозність синдрому може змінитися від помірного до небезпечного для життя.
Синдром названий на честь двох дерматологів, які ідентифікували його на початку XX століття: Едварда Елерса (1863-1937) з Данії та Анрі-Олександра Данлоса (1844-1912).
Клінічні ознаки
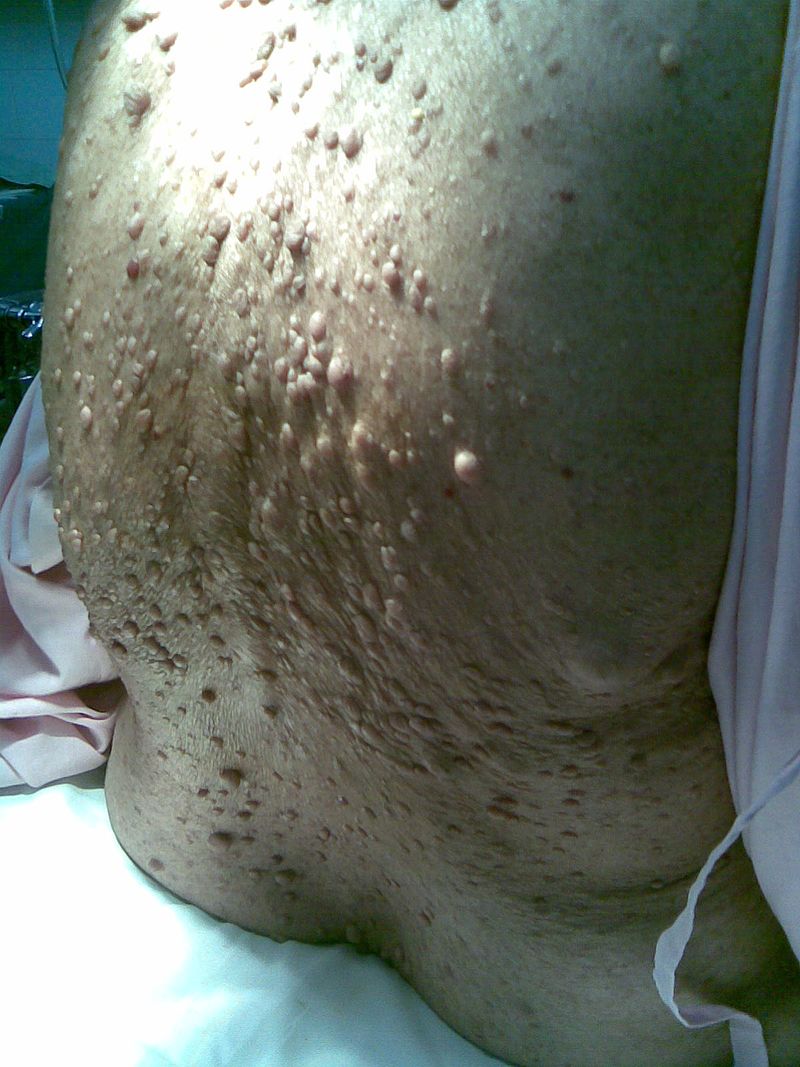
Р ис.8.
- Гіперрухливість суглобів
ис.8.
- Гіперрухливість суглобів
С имптоми
сильно варіюються в залежності від типу
хвороби. Хвороба, як правило, вражає
суглоби, шкіру і кровоносні судини, з
симптомами, такими як вільні, сильно
гнуться суглоби (рис.8); гладка або
еластична, легко пошкоджуються шкіра;
неправильне загоєння ран і формування
шрамів; маленькі і тендітні кровоносні
судини. Всі форми зачіпають суглоби,
викликаючи гіперрухливість, вони
виходять за межі нормального діапазону
рухів. В результаті люди з синдромом
Елерса-Данлоса більш схильні до різних
травм таким як: вивихи, підвивихи,
розтягнення зв'язок, деформація і іноді
розрив м'яких тканин. Так як синдром
часто не діагностується, деякі випадки
приймаються за жорстоке поводження з
дітьми.
имптоми
сильно варіюються в залежності від типу
хвороби. Хвороба, як правило, вражає
суглоби, шкіру і кровоносні судини, з
симптомами, такими як вільні, сильно
гнуться суглоби (рис.8); гладка або
еластична, легко пошкоджуються шкіра;
неправильне загоєння ран і формування
шрамів; маленькі і тендітні кровоносні
судини. Всі форми зачіпають суглоби,
викликаючи гіперрухливість, вони
виходять за межі нормального діапазону
рухів. В результаті люди з синдромом
Елерса-Данлоса більш схильні до різних
травм таким як: вивихи, підвивихи,
розтягнення зв'язок, деформація і іноді
розрив м'яких тканин. Так як синдром
часто не діагностується, деякі випадки
приймаються за жорстоке поводження з
дітьми.
Класифікація
За винятком типу гіперрухливості, були ідентифіковані і ототожнені окремі мутації - вони можуть бути точно визначені генетичним аналізом.
Тип Гіперрухливість
Тип 3 зустрічається у 1 людини на 10 000 - 15 000, що робить його найпоширенішим варіантом хвороби. Ознаки та симптоми можуть бути не діагностовані лікарями або, як правило, помилково діагностовано як фіброміалгія і зазвичай хворим не ставлять діагноз, поки не виявляться серйозні ускладнення. Діагностика здійснюється, в основному, на клінічних спостереженнях. Основні ознаки та симптоми включають в себе:
вільні, нестабільні суглоби, схильні до: розтягування, вивихів, підвивихи (частковий вивих), перерозгинання суглобів
плоскостопість
високе й вузьке піднебіння
легкі синці
легко пошкоджуються оксамитово-гладка шкіра
ранній початок остеопорозу (зазвичай проявляється в середині 30 років)
ураження серця: набута вада серця (така як пролапс мітрального клапана, що створює підвищений ризик інфекції (ендокардит) під час операцій (також можливий розвиток до вкрай небезпечного для життя ступеня при пролапсі мітрального клапана).
Інші симптоми та ускладнення можуть включати в себе:
низька щільність кісток (остеопенія) - попередник остеопорозу
м'язова слабкість, часто ускладнюється холодною погодою
деформації хребта, такі як: сколіоз (викривлення хребта), кіфоз (горб в грудному відділі), en: Tethered spinal cord syndrome, базилярна інвагінація (cranial settling), а також Мальформація Арнольда - Кіарі (ураження довгастого мозку, мозочка виражені потиличним болем, порушеннями ковтання, атаксією та іншими симптомами).
функціональні розлади кишечника (функціональний гастрит, синдром роздратованого кишечника)
здавлення нервів (синдром зап'ястного каналу, парестезія, невралгія трійчастого нерва)
хвороба Рейно
міалгія (біль в м'язах) і артралгії
надмірна втома
передчасний розрив амніону (викидень) під час вагітності en: Premature rupture of membranes
Немовлята з гіперрухливістю суглобів мають слабкий м'язовий тонус (м'язова гіпотонія), який може затримати розвиток таких моторних навичок як самостійне присадка, вставання і ходіння.
Біль, який супроводжує цей стан, є серйозним ускладненням.
Класичний тип
При старій системі класифікації він був розділений на два типи: тип I (важкий) і тип II (помірний). Вражає приблизно від 2 до 5 осіб на 100 000, і є другим за поширеністю. Вражає колаген V і I типу. Важливі симптоми зачіпають шкіру і суглоби. Хворі як правило мають наступні прояви:
гладка, сильно еластична шкіра, легко пошкоджується;
потворні або надзвичайно великі шрами, особливо на лобі, колінах, ліктях і підборідді;
гіперрухливість суглобів, мають тенденцію до вивихів, розтягування зв'язок і підвивихів (зазвичай у колінному суглобі, плечі, п'ястно-фаланговому суглобі і в скронево-щелепному суглобі.
Через зниженого м'язового тонусу, у немовлят може бути порушений розвиток моторних навичок
В даний час не існує певного тесту для діагностики цього типу синдрому. І ДНК аналіз і біохімічні дослідження використовуються для виявлення уражених хворобою. У деяких випадках біопсія шкіри була визнана корисною при постановці діагнозу. На жаль ці тести не досить надійні, щоб виявити всіх хворих. Якщо в родині є кілька хворих членів, то можна провести внутрішньоутробну ДНК діагностику.
Судинний тип
Вражає приблизно 1 людину на 100 000, викликаний аутосомним домінантним дефектом у синтезі колагену типу III. IV тип є найнебезпечнішим різновидом синдрому. Проведені дослідження визначають очікувану тривалість життя приблизно до 48 років. Проте, ця цифра ймовірно спотворена і заснована на факті, що цей тип (як всі інші типи синдрому) істотно не виявляють, і висока пропорція смертельних випадків викликана посмертним діагностуванням. Підвищення обізнаності серед лікарів і населення може допомогти зробити цю цифру більш точною, і скоротити число передчасних смертей.
Ознаки та симптоми:
гіперрухливість, найбільш очевидна на пальцях рук і ніг;
тендітні стінки судин оболонок органів і ніжної шкіри, мають схильність до розриву або утворення аневризми;
хворі як правило мають тонку, бліду і прозору шкіру (можна бачити вени на грудях);
артеріальна / кишкова / маткова крихкість судин або тріщини, розриви;
великі синці.
Деякі пацієнти мають характерні риси обличчя (великі очі, маленьке підборіддя, тонкий ніс і губи, м'які вуха) і мають маленький зріст
В результаті можливості маточного розриву, вагітність може виявитися небезпечною для життя. Доступне лабораторне тестування. Шкірна біопсія може служити доказом аномальної структури колагену. Цей біомеханічний аналіз виявляє більше 95% випадків. Лабораторне тестування рекомендується особам які мають два або більше зазначених симптомів. ДНК аналіз може виявити зміни в межах гена COL3A1. Ця інформація допомагає у передпологовій генетичної консультації, коли один з батьків діагностований і відома його генетична мутація або був продемонстрований біомеханічний дефект.
Тип кифосколіоз
Тип VI дуже рідкісний, виявлено трохи більше 60 випадків, передається аутосомно-рецесивним механізмом. Основним симптом є загальна нестабільність (неміцність) суглобів. У немовлят спостерігається слабкий м'язовий тонус, затримка в розвитку моторних навичок, прогресуюче протягом життя ненормальне викривлення хребта - сколіоз, при якому зазвичай хворі не можуть ходити до 20 років. Легко ранимі очі і шкіра, також можлива вразливість кровоносних судин. Спостерігаються: спонтанне відшарування сітківки, крововиливи у скловидне тіло, розриви очного яблука і рогівки, склер. Також у кісток може бути знижена щільність. Існує чотири основних медичних критерію для діагностики цього типу.
гіперрухливість суглобів,
слабкий м'язовий тонус у новонароджених,
прогресуючий з народження сколіоз,
тендітні (вразливі) очі, що може надати блакитний відтінок склер або викликати розрив очей.
Викликаний змінами в хромосомі 1 гена PLOD, який кодує фермент лізил-гідролази. Можливий лабораторний тест, в ньому вимірюється вміст в сечі hydroxylysyl pryridinoline'а. Він вкрай чутливий і специфічний для даного типу синдрому, рекомендується немовлятам з трьома і більше основними симптомами. Внутрішньоутробний аналіз застосовується, якщо відомо про існуючий ризик: діагностовані хворі члени сім'ї, які мають позитивні результати тестування. Вищезгаданий амніоцентез може бути виконаний, якщо зародкові клітини беруться з амніотичної рідини і виміряна активність ферменту.
Діагностика
• клінічні симптоми;
• історія сім'ї;
• біопсія шкіри (для діагностики судинних, артрохалізаційних та дерматопараксових типів синдрому);
• аналіз сечі (для типу кифосколіозу).
Лікування
Лікування немає, але існує терапія (догляд), яка пом'якшує наслідки хвороби.
Медична терапія спирається на керування симптомами і намагається запобігти подальшим ускладненням.
Великі добові дози (1-4 г) вітаміну С можуть зменшити синці та допомогти ву загоєнні ран. Терапія закликає захищати шкіру сонцезахисним кремом та уникати діяльності, яка створює травму суглобів.
Прогноз
Прогноз для людей з синдромом Елерса-Данлоса залежить від типу. Більшість людей матимуть сприятливий прогноз. Люди з судинними типами мають підвищений ризик фатальних ускладнень.
