

-
7.1. Понятие об энергии активации химической реакции
-
7.2. Основные понятия и определения химической кинетики
-
7.3. Кинетическая классификация гомогенных химических реакций
-
7.4. Способы определения порядка реакции
- 7.5. Влияние температуры на скорость химической реакции
7.1. Понятие об энергии активации химической реакции
На предыдущих лекциях мы рассматривали закономерности химических процессов с точки зрения термохимии. Такой подход позволяет нам на основе изучения энергетических свойств системы и ее энтропии определить тепловой эффект реакции и прогнозировать направление ее протекания. При этом очень часто полученное из термодинамического рассмотрения заключение об осуществимости процесса не означает, что данная химическая реакция в действительности осуществится в рассматриваемых условиях. Например, нас интересует превращение одной кристаллической модификации углерода - алмаза - в другую его модификацию - графит.
- [TEX]С(алмаз)\rightarrow{С(графит)}[/TEX]
Термодинамическое изучение такой системы показывает, что при стандартных условиях графит устойчивее алмаза: Gгр = -1,71 кДж/моль, Gал = 1,18 кДж/моль. Поэтому в указанных условиях алмаз должен самопроизвольно превращаться в графит, т.к. ∆G = Gгр - Gал = -2,89 кДж/моль. На самом деле, как мы знаем, этого превращения не происходит. Такое кажущееся противоречие теоретических расчетов и практических наблюдений объясняется ограниченностью термодинамического метода, т.к. он рассматривает лишь начальное и конечное состояние системы.
Действительно, при обычных условиях свободная энергия графита меньше свободной энергии алмаза. Дело, однако, в том, что превращение кристаллической решетки алмаза в решетку графита обязательно проходит через какие-то промежуточные структуры, отличающиеся от обеих решеток (рис. 7.1).
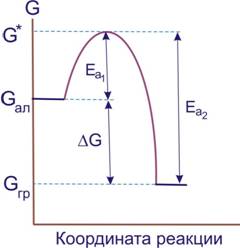
Рисунок 7.1 ‒ К определению энергии активации
Образование промежуточных структур связано с затратой энергии, достигающей G*. Поэтому фактическое изменение энергии в процессе реакции изобразится кривой с максимумом. Высоту максимума по сравнению с энергией исходного вещества называют энергией активации данной химической реакции Еа. Иными словами, термодинамически возможный переход системы из начального состояния в конечное связан с преодолением энергетического барьера, затрудняющего реальное течение процесса, особенно при низких температурах.
Можно привести много примеров, когда термодинамически возможные процессы практически не совершаются из-за ничтожно малых скоростей. Так реакция взаимодействия водорода и кислорода
- [TEX]2H_{2}+O_{2}\rightarrow{2H_{2}O}[/TEX]
сопровождается очень большой убылью энергии Гиббса ∆G298=-455,6кДж/моль, т.е. процесс должен идти самопроизвольно. Однако известно, что смесь этих газов (гремучая смесь) может пребывать сколь угодно долго без заметного образования воды. Дело опять же в высоком энергетическом барьере, в высокой энергии активации. Дадим определение этой величине. Энергия активации - это то избыточное количество энергии (по сравнению со средней величиной), которой должны обладать молекулы в момент столкновения, чтобы прореагировать. Эта избыточная энергия может быть в молекулах в различных формах. Это может быть: 1) повышенная кинетическая энергия поступательного или вращательного движения молекулы; 2) повышенная энергия внутримолекулярных колебаний атомов; 3)повышенная энергия движения электронов и т.д..
Как видно из рис. 7.1, энергии активации прямой (Еа1) и обратной (Еа2) реакции различны. Например, для реакции [TEX]H_{2}+I_{2}=2HI[/TEX] Еа1 = 166,0 кДж/моль, a Ea2 = 185,6 кДж/моль.
Из вышеизложенного ясно, что изучение химических систем с одной лишь термодинамической точки зрения недостаточно. Не менее важно изучение во втором аспекте - с точки зрения скоростей процессов и их механизмов, т.е. с точки зрения кинетики.
7.2. Основные понятия и определения химической кинетики
Скоростью химической реакции W называют изменение количества какого-либо вещества (либо исходного, либо продукта) ni в единицу времени t и в единице реакционного пространства R:
- (7.1)[TEX]W=\pm{\frac{1}{R}\frac{dn_{i}}{dt}}[/TEX]
Это выражение используют со знаком "плюс", когда скорость реакции определяют по изменению количества продукта реакции (которое положительно), или со знаком "минус", когда скорость реакции определяют по изменению количества какого-либо исходного вещества (которое отрицательно). И в том, и в другом случае скорость реакции остается положительной.
Приведенное определение скорости химической реакции является общим, оно справедливо для любых реакций. Если реакция гомогенная и протекает в объеме, то реакционным пространством является объем [TEX](R\equiv{V})[/TEX]. Если же реакция гетерогенная и протекает на поверхности раздела фаз, то реакционным пространством является эта поверхность [TEX](R\equiv{S})[/TEX].
Наиболее теоретически описанными с точки зрения химической кинетики являются гомогенные реакции в изолированных или закрытых системах. Основные закономерности химической кинетики, поэтому, рассмотрим на примере таких систем. Это могут быть либо газовые реакции, либо реакции между растворами (при условии, что реагирующая смесь представляет собой одну фазу). В этом случае для скорости реакции можно записать
- (7.2)[TEX]W=\pm{\frac{1}{V}\frac{dn_{i}}{dt}}[/TEX]
Если V = const, то, учитывая, что ni/V=Ci, где Ci - концентрация какого-либо компонента, можно записать, что
- (7.3)[TEX]W=\pm{\frac{dC_{i}}{dt}}[/TEX]
Решим еще один принципиальный вопрос. Пусть в системе протекает гомогенная реакция с участием веществ А, В, D, F:
- [TEX]aA+bB=dD+fF[/TEX]
И пусть концентрации веществ соответственно равны CA, СB, СD и СF. Тогда для однозначного определения скорости реакции достаточно следить за изменением концентрации одного какого-либо вещества. Изменения концентраций всех остальных участников реакции можно найти из соотношения
- (7.4)[TEX]W=-\frac{dC_{A}}{dt}=-\frac{a}{b}\frac{dC_{B}}{dt}=\frac{a}{d}\frac{dC_{D}}{dt}=\frac{a}{f}\frac{dC_{F}}{dt}[/TEX]
Введем понятие об элементарной реакции. Такой реакцией называется реакция, протекающая в соответствии с ее уравнением, т.е. за счет одновременного столкновения молекул, записанных в левой части уравнения. Следует сказать, что с этих позиций в элементарной реакции могут участвовать одна, две или, что встречается крайне редко, три молекулы. Элементарные реакции поэтому подразделяются на моно-, би- и тримолекулярные.
Математическое уравнение, связывающее скорость реакции с концентрациями реагирующих веществ, называют кинетическим уравнением данной химической реакции. Следует отметить, что по внешнему виду уравнения химической реакции нельзя сделать вывод об ее кинетическом уравнении (напомним, что в химической термодинамике по внешнему виду уравнения можно провести расчет различных величин, например, константы равновесия). Проиллюстрируем сказанное. Реакции водорода с парами йода или брома выражаются одинаковыми стехиометрическими уравнениями. Однако кинетические уравнения для них совершенно различны. Именно:
- [TEX]H_{2}+I_{2}=2HI, \frac{dC_{HI}}{dt}=k\cdot{C_{H_{2}}}\cdot{C_{I_{2}}};[/TEX]
- [TEX]H_{2}+Br_{2}=2HBr, \frac{dC_{HBr}}{dt}=\frac{k\cdot{C_{H_{2}}}\cdot{C_{Br_{2}}^{\frac{1}{2}}}}{1+k'C_{HBr}/C_{Br_{2}}}[/TEX]
Это объясняется тем, что первая реакция протекает как элементарная, в то же время образование HBr происходит в результате сложной ценной реакции, механизм которой можно представить так:
Hr2⇄2Br
Br+H2⇄HBr+H
H+Br⇄HBr+Br
Таким образом, кинетическое уравнение химической реакции может быть установлено исключительно экспериментальным путем.
Основным законом химической кинетики является постулат, вытекающий из большого числа экспериментальных данных и выражающий зависимость скорости от концентрации. Этот закон, получивший название закона действующих масс, формулируется следующим образом.
Если эмпирическое уравнение реакции
- [TEX]aA+bB+dD\rightarrow{продукты}[/TEX]
то математическую формулу основного закона можно представить в виде
- (7.5)[TEX]W=-\frac{dC_{A}}{dt}=kC_{A}^{n_{A}}\cdot{C_{B}^{n_{B}}}\cdot{C_{D}^{n_{D}}}[/TEX]
В этом уравнении коэффициент пропорциональности k не зависит от концентраций. Он очень различен для разных реакций и сильно зависит от температуры. Называют k константой скорости реакции. Физический смысл k легко найти положив СA = CB = СD = 1 моль/л. Тогда W=k, т.е. k численно равна скорости реакции при концентрациях реагирующих веществ, равных единице. Показатели степеней nA, nB, nD обычно являются положительными целыми числами, но бывают дробные и даже отрицательные показатели. Появление этих усложнений объясняется более или менее сложным механизмом реакции.
7.3. Кинетическая классификация гомогенных химических реакций
С точки зрения химической кинетики все элементарные реакции можно классифицировать на реакции нулевого, первого, второго и третьего порядка. В отличие от молекулярности порядок реакции величина формальная и равная сумме показателей степеней в кинетическом уравнении. Из соотношения (7.5) общий порядок n данной реакции равен
- [TEX]n=n_{A}+n_{B}+n_{D}[/TEX]
При этом величины nA, nB, nD называются частными порядками реакции по веществам А, В и D. Следовательно, общий порядок реакции равен сумме частных порядков.
Для элементарных реакций nA = a, nB = b, nD = d. Возможны следующие типы кинетических уравнений элементарных реакций:
| [TEX]n=1[/TEX] | [TEX]A\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A}[/TEX] |
| [TEX]n=2[/TEX] | [TEX]2A\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A}^{2}[/TEX] |
| [TEX]A_{1}+A_{2}\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A_{1}}\cdot{C_{A_{2}}}[/TEX] | |
| [TEX]n=3[/TEX] | [TEX]3A\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A}^{3}[/TEX] |
| [TEX]2A_{1}+A_{2}\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A_{1}}^{2}\cdot{C_{A_{2}}}[/TEX] | |
| [TEX]A_{1}+A_{2}+A_{3}\rightarrow{продукты}[/TEX] | [TEX]W=kC_{A_{1}}\cdot{C_{A_{2}}}\cdot{C_{A_{3}}}[/TEX] | |
| [TEX]n=0[/TEX] | [TEX]A\rightarrow{продукты}[/TEX] | [TEX]W=k[/TEX] |
Реакции нулевого порядка это реакции, которые протекают в избытке реагентов так, что изменение концентрации не влияет на скорость реакции.
Найдем основные кинетические характеристики и уравнения для гомогенных химических реакций различного порядка в закрытых системах при постоянстве объема и температуры.
- Реакции нулевого порядка. Такие реакции редки. Примером реакции нулевого порядка является реакция разложения N2O5 в газовой фазе в присутствии твердого N2O5, когда постоянная концентрация реагента в газовой фазе поддерживается постоянной за счет испарения твердого оксида. Для реакций нулевого порядка
- (7.6)[TEX]W=-\frac{dC}{dt}=k[/TEX]
Решим это уравнение, разделив переменные
- [TEX]dC=-kdt[/TEX]
После интегрирования [TEX]\int{dC}=-\int{kdC}[/TEX] получим
- [TEX]C=-kt+B[/TEX]
где В - константа интегрирования. Найдем ее, учитывая начальное условие: в момент t=0 концентрация равна начальной С = С0. Отсюда
- (7.7)[TEX]C=C_{0}-kt[/TEX]
Следовательно, в реакциях нулевого порядка концентрация линейно уменьшается со временем (рис. 7.2, а).
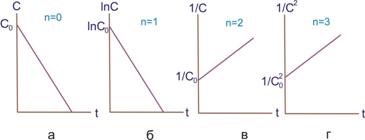
Рисунок 7.2 ‒ Изменение концентрации со временем в реакциях нулевого (а), первого (б), второго (в) и третьего (г) порядков
Константу скорости реакции нулевого порядка можно вычислить по уравнению
- (7.8)[TEX]k=\frac{1}{t}(C_{0}-C)[/TEX]
Ее размерность (моль/л)с-1.
Наряду с константой скорости для характеристики реакций часто пользуются величиной, называемой временем полупревращения [TEX]\tau_{1/2}[/TEX]. Время полупревращения - промежуток времени, в течение которого реагирует половина исходного количества вещества, т.е. при [TEX]t=\tau{_{1/2}}[/TEX] [TEX]C=\frac{C_{0}}{2}[/TEX]. Отсюда
- [TEX]\frac{C_{0}}{2}=C_{0}-k\tau_{_{1/2}}[/TEX]
- (7.9)[TEX]\tau_{_{1/2}}=\frac{C_{0}}{2k}[/TEX]
Следовательно, для реакции нулевого порядка время полупревращения пропорционально начальной концентрации исходного вещества.
- Реакции первого порядка. К элементарным реакциям первого порядка относятся реакции, которые можно представить в виде:
- [TEX]A\rightarrow{продукты}[/TEX]
Чаще всего это реакции разложения. Например, реакция разложения ацетона
- [TEX]CH_{3}COCH_{3}\rightarrow{CH_{4}+CO+H_{2}}[/TEX]
Кинетическое уравнение для реакций первого порядка имеет вид
- (7.10)[TEX]W=-\frac{dC}{dt}=kC[/TEX]
Проинтегрируем его, разделив переменные
- [TEX]\int{\frac{dC}{C}}=-\int{kdt}[/TEX]
- [TEX]\ln{C} =-kt+B[/TEX]
Для определения постоянной интегрирования примем, что в начальный момент реакции t=0 концентрация исходного вещества была С0. Тогда
- [TEX]\ln{C_{0}}=B[/TEX]
Следовательно, [TEX]\ln{C}=-kt+\ln{C_{0}} [/TEX] или
- [TEX]\ln{\frac{C}{C_{0}}}=-kt[/TEX]
- (7.11)[TEX]C=C_{0}e^{-kt}[/TEX]
Это интегральная форма кинетического уравнения реакции 1-го порядка. Константа скорости реакции первого порядка имеет размерность (время)-1. Величина, обратная константе скорости реакции первого порядка имеет размерность времени и называется средней продолжительностью жизни отдельной частицы.
Для реакций первого порядка характерна линейная зависимость lnC от t (рис. 7.2, б).
Найдем время полупревращения для реакции первого порядка. Для [TEX]t=\tau{_{1/2}}[/TEX] С = С0/2.
Поэтому
- [TEX]\ln{\frac{C}{C_{0}}}=\ln{\frac{\frac{C_{0}}{2}}{C_{0}}}=-k\tau{_{1/2}}[/TEX]
- [TEX]k\tau{_{1/2}}=\ln{2}[/TEX]
Отсюда
- (7.12)[TEX]\tau{_{1/2}}=\frac{\ln{2}}{k}=\frac{0.693}{k}[/TEX]
Видно, что время полупревращения определяется исключительно значением константы скорости. Так, для приведенной реакции разложения ацетона k = 4,27ּ10-4 сек-1. Следовательно
- [TEX]\tau{_{1/2}}=\frac{0.693}{4.27\cdot{10^{-4}}}=1580с=26мин20с.[/TEX]
К истинно мономолекулярным или, лучше сказать, моноатомным процессам первого порядка относятся все многочисленные превращения радиоактивных веществ. В табл. 7.1 приведены некоторые данные для таких реакций.
Таблица 7.1 - Кинетические характеристики радиоактивного распада некоторых изотопов
Изотоп
k, c-1
[TEX]\tau_{1/2}[/TEX]
222Ra
226Ra
238U
214Po
2,1×10-6
1,35×10-11
4,88×10-18
4,62×103
3,8 суток
1620 лет
4,5×109 лет
1,5×10-4 с
Классическим примером реакции первого порядка является реакция инверсии тростникового сахара (сахарозы)
Кинетическое уравнение этой реакции имеет вид
- [TEX]-\frac{dC_{сах}}{dt}=kC_{сах}[/TEX]
В реакции участвуют еще вода и кислота (ионы Н+), однако концентрация кислоты постоянна (ионы Н+ - катализатор), а концентрация воды, присутствующей в очень большом избытке, тоже практически постоянна. Реакция инверсии удобна для изучения тем, что в ходе ее изменяется угол вращения плоскости поляризации. Сахароза вращает плоскость поляризации вправо, а смесь глюкозы и фруктозы - влево. Помещая раствор сахарозы в трубку поляриметра, можно следить за ходом реакции, не прерывая ее. При кинетических расчетах используется пропорциональная связь между углом вращения и концентрацией вращающего вещества.
- Реакции второго порядка. Реакции второго порядка протекают с участием двух частиц (молекул, атомов). Если в элементарной реакции участвуют две одинаковые частицы
- [TEX]2A\rightarrow{продукты}[/TEX]
то кинетическое уравнение имеет вид
- (7.13)[TEX]-\frac{dC}{dt}=kC^{2}[/TEX]
где С - концентрация вещества А. Интегрирование этого уравнения приводит к
- [TEX]\frac{1}{С}=kt+B[/TEX]
При t = 0 С = C0. Отсюда [TEX]B=\frac{1}{C_{0}}[/TEX], следовательно
- (7.14)[TEX]\frac{1}{C}-\frac{1}{C_{0}}=kt[/TEX]
или
- (7.15)[TEX]k=\frac{1}{t}\cdot{\frac{C-C_{0}}{C_{0}C}}[/TEX]
Для реакций второго порядка наблюдается линейная зависимость 1/с от t (рис. 7.2, б).
Размерность константы скорости реакции второго порядка (концентрация)-1ּ(время)-1, например, (моль/л)-1ּс-1, т.е. в отличие от реакций первого порядка в размерности есть не только время, но и концентрация. Поэтому для реакций второго порядка (и выше) нельзя при расчетах заменять концентрацию на пропорциональные ей величины.
Для реакции второго порядка
- (7.16)[TEX]\tau_{1/2}=\frac{1}{kC_{0}}[/TEX]
т.е. чем больше начальная концентрация, тем меньше время полупревращения.
Если в элементарной реакции второго порядка реагируют две различные частицы
- [TEX]A_{1}+A_{2}\rightarrow{продукты}[/TEX]
причем концентрации веществ А1 и А2 разные, то
- (7.17)[TEX]W=-\frac{dC_{1}}{dt}=-\frac{dC_{2}}{dt}=kC_{1}\cdot{C_{2}}[/TEX]
Учитывая, что C1 = C10 - X и C2 = C20 - X, имеем
- [TEX]\frac{dX}{dt}=k(C_{10}-X)(C_{20}-X)[/TEX]
где C10 и C20 - начальные концентрации веществ А1 и А2, X - уменьшение их концентрации к моменту времени t. Решение последнего уравнения дает
- (7.18)[TEX]k=\frac{1}{t(C_{10}-C_{20})}\ln{\frac{C_{20}(C_{10}-X)}{C_{10}(C_{20}-X)}} [/TEX]
Реакций второго порядка довольно много. Например:
- [TEX]H_{2}+I_{2(пар)}\rightarrow{2HI}[/TEX]
- [TEX]2HI\rightarrow{H_{2}+I_{2(пар)}}[/TEX]
- Реакции третьего порядка. Элементарные реакции третьего порядка можно представить в виде
- [TEX]3A\rightarrow{продукты}[/TEX]
- [TEX]2A_{1}+A_{2}\rightarrow{продукты}[/TEX]
- [TEX]A_{1}+A_{2}+A_{3}\rightarrow{продукты}[/TEX]
Так как реакции третьего порядка очень редки и не представляют большого практического интереса, рассмотрим их кинетику только для случая равенства концентраций всех реагирующих веществ.
Тогда
- (7.19)[TEX]-\frac{dC}{dt}=kC^{3}[/TEX]
и после интегрирования
- [TEX]\frac{1}{2C^{2}}=kt+B[/TEX]
При t=0, C=C0 и B=1/2C02. Следовательно,
- (7.20)[TEX]\frac{1}{C^{2}}-\frac{1}{C_{0}^{2}}=2kt[/TEX]
или
- (7.21)[TEX]k=\frac{1}{2t}\left(\frac{1}{C^{2}}-\frac{1}{C_{0}^{2}}\right)[/TEX]
Константа скорости реакции третьего порядка имеет размерность (концентрация)-2(время)-1, например, (моль/л)-2ּс-1.
Для реакций третьего порядка линейна зависимость 1/С2=f(t) (рис. 7.2, г) . Время полупревращения для реакций третьего порядка обратно пропорционально квадрату концентрации
- (7.22)[TEX]\tau_{1/2}=\frac{3}{2kC_{0}^{2}}[/TEX]
Примерами реакций третьего порядка являются
- [TEX]2NO+O_{2}\rightarrow{2NO_{2}},[/TEX]
- [TEX]2NO+Cl_{2}=2NOCl[/TEX]
Следует отметить, что реакции первого, второго и третьего порядка являются в то же время моно-, би- и тримолекулярными, соответственно.
Для моделирования химико-технологических процессов полезно ввести понятие формально простых реакций. К формально простым реакциям относятся любые сложные реакции, для которых кинетическое уравнение может быть представлено приближенно в виде степенной зависимости
- [TEX]W=kC_{1}^{n_{1}}\cdot{C_{2}^{n_{2}}}\cdot{C_{3}^{n_{3}}}...[/TEX]
Например, такой является сложная реакция
- [TEX]3CH_{3}OH+2H_{2}CrO_{4}+6HCl\rightarrow{3CH_{2}O+2CrCl_{3}+8H_{2}O}[/TEX]
Для этой реакции
- [TEX]W=kC_{CH_{3}OH}\cdot{C_{H_{2}CrO_{4}}}\cdot{C_{HCl}^{2}}[/TEX]
Как видно, показатели степеней для формально простой реакции не совпадают со стехиометрическими коэффициентами. В общем случае для таких реакций показатели степеней могут быть дробными и принимать значения большие трех.
При равенстве концентраций реагирующих веществ кинетическое уравнение имеет вид
- (7.23)[TEX]-\frac{dC}{dt}=kC^{n}[/TEX]
Решение этого уравнения аналогично предыдущим и приводит к следующим результатам
- (7.24)[TEX]\frac{1}{(n-1)}\left(\frac{1}{C^{n-1}}-\frac{1}{C_{0}^{n-1}}\right)=kt[/TEX]
- (7.25)[TEX]\tau_{1/2}=\frac{2^{n-1}-1}{(n-1)kC_{0}^{n-1}}[/TEX]
Константа скорости имеет размерность (моль/л)-(n-1) × (с)-1.
7.4. Способы определения порядка реакции
Порядок реакции, в котором участвует одно вещество, можно найти, воспользовавшись данными экспериментов для двух различных начальных концентраций С01 и С02. Тогда
- [TEX]W_{01}=-\frac{dC_{01}}{dt}=kC_{01}^{n}[/TEX]
- [TEX]W_{02}=-\frac{dC_{02}}{dt}=kC_{02}^{n}[/TEX]
Логарифмируем эти уравнения и получаем
- [TEX]\ln{W_{01}}=\ln{k}+n\ln{C_{01}}[/TEX]
- [TEX]\ln{W_{02}}=\ln{k}+n\ln{C_{02}}[/TEX]
Вычитаем уравнения друг из друга и получаем
- [TEX]n=\frac{\ln{\frac{W_{01}}{W_{02}}}}{\ln{\frac{С_{01}}{С_{02}}}}[/TEX]
Для реакций, в которых участвуют два исходных вещества
- [TEX]aA+bB\rightarrow{продукты}[/TEX]
кинетическое уравнение имеет вид
- [TEX]W=kC_{A}^{n_{A}}\cdot{C_{B}^{n_{B}}}[/TEX]
и задача сводится к определению частных порядков реакции nA и nB, n = nA + nB.
Для этого проводят две серии экспериментов: один при постоянной (или избыточной) концентрации вещества А, второй при постоянной (или избыточной) концентрации вещества В. В обеих случаях концентрация одного из веществ не будет оказывать влияния на скорость реакции (точнее это влияние будет постоянным). Имеем
- [TEX]W_{1}=-\frac{dC_{B}}{dt}=k_{B}C_{B}^{n_{B}},[/TEX]
- [TEX]W_{1}=-\frac{dC_{A}}{dt}=k_{A}C_{A}^{n_{A}},[/TEX]
где [TEX]k_{B}=kC_{A}^{n_{A}} и k_{A}=kC_{B}^{n_{B}}[/TEX]. После логарифмирования получаем
- [TEX]\ln{W_{1}}=\ln{k_{B}}+n_{B}\ln{C_{B}}[/TEX]
- [TEX]\ln{W_{2}}=\ln{k_{A}}+n_{A}\ln{C_{A}}[/TEX]
Экспериментальные точки наносим на график в координатах lnW – lnС. Получаем две прямые (рис.7.3).
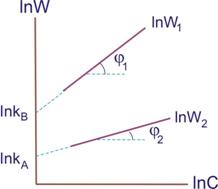
Рисунок 7.3 ‒ К определению порядка реакции
- [TEX]n_{B}=tg\varphi_{1}[/TEX]
- [TEX]n_{A}=tg\varphi_{2}[/TEX]
- [TEX]n=tg\varphi_{1}+tg\varphi_{2}[/TEX]
Величины отрезков, отсекаемых прямыми, на оси ординат позволяют найти k.
Описанные способы называются дифференциальными. Кроме того, существуют интегральные способы определения порядка реакции по экспериментальным данным. Они основаны на проверке выполнения кинетических зависимостей для реакций разных порядков. Рассмотрим эти способы.
- Графический способ. Как видно из рис. 7.3 и проведенных нами решений кинетических уравнений для реакций разных порядков линейная зависимость f(C)-t реализуется в разных координатах. Для нахождения порядка реакции строят все четыре зависимости (C-t; lnC-t; 1/C - t; 1/С2 - t). Если, например, график, построенный по опытным данным, оказался прямолинейным в координатах 1/С – t, то порядок реакции по рассматриваемому веществу равен двум.
- Способ подстановки. Для различных моментов времени по формулам для реакций возможных порядков рассчитывают значения константы скорости k. Если рассчитанные по одной из формул значения k не зависят от времени, то это означает, что эта формула соответствует экспериментально изучаемой реакции.
- Способ времени полупревращения. Последовательно вычисляют [TEX]\tau_{1/2}[/TEX] для нескольких концентраций по формулам для реакций различных порядков, чтобы установить зависимость [TEX]\tau_{1/2}[/TEX] от начальной концентрации. Характер этой зависимости говорит о порядке реакции.
7.5. Влияние температуры на скорость химической реакции
Как известно, скорость большинства химических реакций, за чрезвычайно редким исключением [TEX](2NO+O_2\rightarrow{2NO_2})[/TEX], быстро увеличивается с повышением температуры. Именно поэтому нагревание представляет собой приём, широко распространенный в химической практике. Количественно оценить влияние температуры на скорость (а фактически на константу скорости) реакции можно несколькими путями. Для реакций в растворах, протекающих при сравнительно низких температурах, можно использовать полуколичественное эмпирическое правило Вант-Гоффа:
при повышении температуры на 10° скорость реакции возрастает в 2 - 4 раза.
Введя понятие температурного коэффициента скорости реакции [TEX]\gamma[/TEX], можно рассчитать изменение скорости при любом изменении температуры:
- [TEX]\gamma^n=\frac{k_{(T+10n)}}{k_T}[/TEX]. (7.26)
- Здесь n - число десятков градусов, на которое изменилась температура. n может быть целым и дробным, положительным и отрицательным.
Более точно отражает зависимость константы скорости реакции от температуры уравнение, предложенное в 1889 г. шведским ученым С.Аррениусом (уравнение Аррениуса):
- [TEX]k_T=k_0\exp{\left(-E_a/RT \right)}[/TEX], (7.27)
- где [TEX]k_0[/TEX] - предэкспоненциальный множитель, а [TEX]E_a[/TEX] - энергия активации реакции. Согласно теории Аррениуса (теории активных столкновений) для протекании реакции необходимо:
- чтобы произошли столкновения частиц системы;
- чтобы столкновения частиц действительно привели к химическому превращению, поскольку эффективны не все столкновения, а только те, в которых участвуют "активные" молекулы, имеющие энергию больше [TEX]E_a[/TEX].
Проиллюстрируем важность этого положения простым расчетом. Константа скорости реакции [TEX]2HI\leftrightarrow{I_2+H_2}[/TEX] при [TEX]100^oC[/TEX] равна [TEX]k=8,83\cdot{10^{-16}}(моль/л)^{-1}\cdot{c^{-1}}[/TEX]. Чтобы сделать наглядным это значение м рассчитаем время полупревращения для этой реакции второго порядка (см. 7.3) при С0 = 1 моль/л:
- [TEX]\tau_{1/2}=\frac{1}{kC_0}=\frac{10^{16}}{8,83\cdot{3600}\cdot{24}\cdot{365}}=3,6\cdot{10^7}[/TEX]лет.
Таким образом при [TEX]100^oC[/TEX] половина исходного йодистого водорода разложилось бы за 36 миллионов лет. Причина этого в том, что при данной температуре чрезвычайно мала доля активных молекул. Таблица 7.2 иллюстрирует зависимость доли активных молекул от температуры для различных значений энергии активации.
Таблица 7.2 - Доля активных молекул
|
Т, К |
Энергия активации Еа,Кдж/моль |
|||
|
5 |
50 |
100 |
150 |
|
|
300 600 1000 1500 |
0,13 0,37 0,55 0,67 |
[TEX]2\cdot{10^{-9}}[/TEX] [TEX]4\cdot{10^{-5}}[/TEX] [TEX]2\cdot{10^{-3}}[/TEX] [TEX]2\cdot{10^{-2}}[/TEX] |
[TEX]4\cdot{10^{-18}}[/TEX] [TEX]2\cdot{10^{-9}}[/TEX] [TEX]6\cdot{10^{-6}}[/TEX] [TEX]3\cdot{10^{-4}}[/TEX] |
[TEX]8\cdot{10^{-27}}[/TEX] [TEX]9\cdot{10^{-14}}[/TEX] [TEX]1\cdot{10^{-8}}[/TEX] [TEX]6\cdot{10^{-6}}[/TEX] |
Чем больше Еа, тем сильнее скорость реакции зависит от температуры. Однако, если доля активных молекул больше 10-7, то такая реакция протекает неизмеримо быстро (взрыв), а если меньше 10-18, то реакция идет неизмеримо медленно.
Опыт показывает, что не все столкновения активных молекул приводят к химическим превращениям. Теория активных столкновений объясняет это тем, что взаимодействующие молекулы при столкновении должны быть определенным образом ориентированы в пространстве, т.е. должны образовать конфигурацию, наиболее подходящую для разрыва одних связей и возникновения других. Уравнение Аррениуса примет вид:
- [TEX]k=A\cdot{Pe^{-E_a/RT}}[/TEX], (7.28)
Здесь Р - стерический множитель, определяющий пространственную ориентацию взаимодействующих молекул ([TEX]P\leq 1[/TEX]); А - постоянная ([TEX]A\cdot{P}=k_0[/TEX]). Шло показано, что [TEX]P=\exp{(S_a/R)} [/TEX], где [TEX]S_a[/TEX]- энтропия активации. Поэтому
- [TEX]k=Z\cdot{e^{-E_a/RT}}\cdot{e^{S_a/R}}[/TEX], (7.29)
- где Z - постоянная. Можно говорить, следовательно, об энтальпийном ([TEX]\exp{[-E_a/RT]}[/TEX]) и энтропийном ([TEX]\exp{S_a/R}[/TEX]) факторах. Однако энтропийный фактор не зависит от температуры. Его роль особенно заметна для реакций, в которых участвуют сложные молекулы, взаимная ориентация которых должна быть такой, чтобы друг к другу подошли определенные участки взаимодействующих молекул. Например, для реакции
- [TEX]H_2+I_2\rightarrow{2HI}[/TEX]
P ~ 1, а для реакции
- [TEX]C_2H_5I+(C_2H_5)_3N\rightarrow{(C_2H_5)_4NI}[/TEX]
P ~ 10-6.
Для нахождения констант уравнения Аррениуса (7.27) данные определения скорости реакции при различных температурах представляют в логарифмическом виде:
- [TEX]\ln{k}=\ln{k_0}-\frac{E_a}{RT} [/TEX]
Затем в координатах [TEX]\ln{k}-1/T[/TEX] строят график прямой (рис. 7.4), тангенс угла наклона которой равен [TEX]-E_a/R[/TEX], а отрезок, отсекаемый продолжением прямой на оси ординат, равен [TEX]\ln{k_0}[/TEX] (удобнее рассчитать уравнение этой прямой на ЭВМ и сразу получить ее коэффициенты).
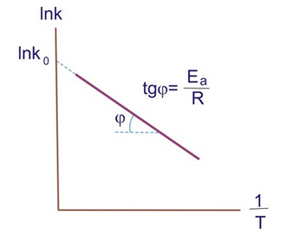
Рисунок 7.4‒ Графическое представление энергии активации
Следует отметить, что хотя скорость большинства реакций растет с повышением температуры, этот рост не всегда монотонный и иногда на кривых зависимости скорости от температуры наблюдаются максимумы или минимумы. Это связано со сложным механизмом таких реакций, наличием катализаторов и рядом других причин.

