

- 5.1 Загальні поняття та величини хімічної термодинаміки
- 5.2 Внутрішня енергія. Перший закон термодинаміки
- 5.3 Ентальпія. Тепловий ефект реакції
- 5.4 Основи термохімії
- 5.5 Ентропія. Другий закон термодинаміки
- 5.6 Напрям перебігу хімічних реакцій. Енергія Гіббса
- 5.7 Приклади розв'язання типових задач
Ключові терміни:
Інтенсивні параметри, Внутрішня енергія, Другий закон термодинаміки, Другий наслідок закону Гесса, Екстенсивні параметри, Енергія Гіббса, Енергія Гіббса хімічної реакції, Ентальпія, Ентропія, Змінення ентропії, Перший закон термодинаміки, Перший наслідок закону Гесса, Принцип самочинного перебігу хімічних реакцій, Робота, Самочинні процеси, Система, Стан системи, Стандартні умови, Теплота, Термодинамічні параметри, Термохімічні рівняння, Термохімія, Третій закон термодинаміки, Фаза, Хімічна термодинаміка, внутрішня енергія, відкрита система, гетерогенна система, гомогенна система, енергія Гіббса, енергія Гіббса утворення, ентальпійний фактор, ентальпія, ентальпія утворення, ентальпія утворення речовини, ентальпія хімічної реакції, ентропійний фактор, ентропія, закон Гесса, закрита система, критерій самочинного перебігу хімічної реакції, нерівноважний стан, перший закон термохімії, рівноважний стан, сполуки, стала Больцмана, стандартна енергія Гіббса, стандартна енергія Гіббса хімічної реакції, стандартна ентальпія утворення, стандартна ентальпія хімічної реакції, стандартна ентропія, стандартні стани речовин, тепловий ефект, тепловий ефект хімічної реакції, термодинамічна імовірність, термодинамічний процес, функція стану, характеристична функція, ізольована система5.1 Загальні поняття та величини хімічної термодинаміки
Хімічна термодинаміка – розділ хімії, який вивчає енергетичні ефекти, що супроводжують хімічні процеси, а також напрямок та межі їх самочинного перебігу. Об’єктами вивчення термодинаміки є система, яка перебуває у певному енергетичному стані та має певний фазовий склад, і термодинамічний процес.
Система – це сукупність взаємодіючих речовин, які уявно або фактично відокремлені від навколишнього середовища.
Фаза – це гомогенна частина системи, що характеризується однаковими фізичними і хімічними властивостями та складом і відокремлюється від інших частин системи поверхнею поділу, при переході через який відбувається стрибкоподібне змінювання властивостей.
Термодинамічні системи (або просто системи) прийнято класифікувати за різними ознаками.
-
За характером взаємодії з навколишнім середовищем системи бувають:

Рисунок 5.1 – Приклади умовно ізольованих систем

Рисунок 5.2 – Приклади закритих систем

Рисунок 5.3 – Приклади відкритих систем
- ізольована система, в якій відсутні енергообмін і масообмін з навколишнім середовищем. Зазвичай вважається, що абсолютно ізольованих систем у земних умовах не існує, але у грубому наближені як приклад ізольованої системи можна навести термос (рис. 5.1) – за умови, що спостереження за ним ведеться дуже короткий проміжок часу;
- закрита система, що обмінюється з навколишнім середовищем лише енергією (рис. 5.2);
- відкрита система, або незамкнута, яка обмінюється з навколишнім середовищем і речовиною і енергією (рис. 5.3).
-
За фазовим складом системи поділяються на:
- гомогенна система, що містить тільки одну фазу, наприклад, суміш газів, однорідний розплав солей чи розчин (рис. 5.4);
- гетерогенна система, яка складаються з декількох фаз, відокремлених одна від одної поверхнею поділу (рис. 5.5), наприклад, лід і рідка вода, рідина та її пара, дві рідини, що не змішуються між собою: вода і гас.

Рисунок 5.4 – Приклади гомогенних систем: а) суміш газів; б) водні розчини індивідуальних солей

Рисунок 5.5 – Приклади гетерогенних систем: а) рідина-газ; б) рідина-рідина; в) газ-тверда фаза; г) рідина-тверда фаза
Фазовий стан неприпустимо плутати з агрегатним станом. Ці поняття співпадають тільки для газів (наприклад, СО і СО2, N2 і NO), які повністю змішуються завдяки високій кінетичній енергії газових молекул та їх невпорядкованому руху. З рідким агрегатним станом справа виглядає інакше. При змішуванні водних розчинів речовин, що не взаємодіють між собою (наприклад КОН і NaOH), вони складатимуть одну рідку фазу і будуть гомогенною системою. З іншого боку, вода і рослинна олія, хоч і перебувають в одному агрегатному стані, але уявляють собою окремі фази, що мають різний склад, різні фізичні та хімічні властивості і – головне – відділяються одна від одної межею поділу. Те ж саме стосується і твердого агрегатного стану. Так, якщо міцно притиснути один до одного графіт і алмаз – дві алотропні модифікації карбону, вони не стануть однією фазою, оскільки мають поверхню поділу і різні кристалічні структури та, як наслідок, відрізняються за своїми властивостями.
Стан системи описується за допомогою фізичних величин, які називаються термодинамічними параметрами.
Термодинамічні параметри – це певні характеристики, що мають важливі відмінні ознаки: змінювання їх величин приводить до зміни стану всієї системи.
Термодинамічні параметри, як і більшість фізичних величин, можуть бути інтенсивними чи екстенсивними. Інтенсивні параметри – це характеристики, значення яких не залежить від розміру системи, наприклад, температура, густина, концентрація, тиск. При розділі системи на декілька підсистем, у кожній зберігається однакове значення термодинамічного параметру. Екстенсивні параметри – це ті термодинамічні параметри, що залежать від розміру системи. Їх значення є адитивною величиною, оскільки складається з відповідних значень окремих підсистем, наприклад: маса, об’єм, кількість речовини, потужність.
- рівноважний стан, якщо термодинамічні параметри однакові в усіх точках системи і не змінюються самочинно протягом часу;
- нерівноважний стан, якщо термодинамічні параметри з часом змінюються самочинно, тобто без витрати енергії (або виконання роботи) ззовні.
Перехід системи із одного стану в інший, при якому змінюються термодинамічні параметри,– це термодинамічний процес.
Щоб не порушувалася термодинамічна рівновага системи з навколишнім середовищем, процес повинний здійснюватися дуже повільно, а в ідеалі – нескінченно довго. При цьому можуть змінюватися всі або окремі параметри системи. Залежно від сталості певних параметрів термодинамічні процеси поділяються на типи (рис. 5.6):
- ізобаричні (Р = const);
- ізохоричні (V = const);
- ізотермічні (Т = const);
- адіабатичні (Q=const).
При сталості двох параметрів процес належить до комбінованих; це відбивається в його назві, наприклад, ізобарно-ізотермічний процес, якщо Р,Т = const.
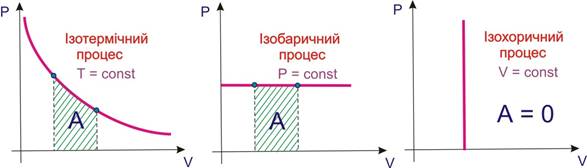
Рисунок 5.6 – Термодинамічні процеси: ізотермічний; ізобаричний; ізохоричний
Термодинамічні властивості системи виражаються за допомогою спеціальних функція стану, або характеристична функція, які мають дві основні особливості:
- їх значення не залежать від способу (або шляху) досягнення даного стану системи, а визначаються лише станом самої системи, який описується за допомогою термодинамічних параметрів. Тому змінення термодинамічних функцій дорівнює різниці між значеннями термодинамічних параметрів у кінцевому і вихідному станах системи (змінення позначають грецькою буквою[TEX]\Delta[/TEX]–дельта);
- значення характеристичних функцій залежать від кількості (або маси) речовини, тому їх відносять до одного моля речовини.
Найчастіше застосовуються такі характеристичні функції: внутрішня енергія U, ентальпія Н, ентропія S та енергія Гіббса G.
5.2 Внутрішня енергія. Перший закон термодинаміки
Хімічні реакції супроводжуються виділенням або поглинанням енергії у вигляді теплоти, світла, випромінювання, роботи тощо. Виділення енергії внаслідок взаємодії речовин доводить, що у них ця енергія існувала у прихованій формі ще до початку реакції. Така прихована енергія, яка звільнюється під час хімічних реакцій і при фізичних явищах (конденсація пари, кристалізація рідин) являє собою внутрішню енергію.
Внутрішня енергія – це функція стану, яка складається з усіх видів енергії системи (енергії руху та взаємодії молекул, атомів, ядер та інших частинок), за винятком кінетичної енергії руху системи як єдиного цілого і потенціальної енергії її положення.
Внутрішня енергія залежить тільки від стану системи, тому неможливо виміряти її абсолютне значення, однак можна встановити її змінення[TEX]\Delta{U}[/TEX]при переході системи з одного стану в інший:
- [TEX]\Delta{U}=U_2-U_1[/TEX]
- де U2 і U1 – відповідно внутрішня енергія у кінцевому і початковому станах. Вимірюється внутрішня енергія у [кДж/моль].
Значення[TEX]\Delta{U}[/TEX]додатне[TEX](\Delta{U}>0)[/TEX], якщо внутрішня енергія системи зростає (U2 > U1), і від’ємне[TEX](\Delta{U}<0)[/TEX]при зменшенні внутрішньої енергії системи (U2 < U1).
Між термодинамічною системою та навколишнім середовищем може відбуватися обмін енергією у вигляді теплоти і роботи.
Теплота Q – це енергія, що передається від одного тіла до іншого при безпосередньому контакті і залежить тільки від їх температур, але не пов’язана з перенесенням речовини.
Теплота, одержана системою, називається підведеною і вважається додатною (Q>0). І навпаки, віддана системою теплота називається відведеною і вважається від’ємною (Q<0).
Теплота Q є кількісною мірою хаотичного руху частинок даної системи, а робота А –кількісною мірою напрямленого руху частинок, або мірою енергії, що передається від однієї системи до іншої за рахунок переміщення речовини під дією певних сил (наприклад, гравітаційних).
Робота А – це енергія, що передається одним тілом іншому при їх взаємодії, не залежить від температури цих тіл і не пов’язана з передаванням теплоти.
Додатною вважаються робота (А>0), що виконується системою проти дії зовнішніх сил, а від’ємною (А<0) – робота, яку навколишнє середовище виконує щодо системи.
Теплота і робота вимірюються у кілоджоулях [кДж].
На відміну від внутрішньої енергії U теплота Q і робота A залежать від способу проведення процесу, тому вони не належать до характеристичних функцій.
Співвідношення між зміненням внутрішньої енергії[TEX]\Delta{U}[/TEX], теплотою Q і роботою A встановлює
теплота, підведена до системи, витрачається на збільшення внутрішньої енергії системи і на її роботу над навколишнім середовищем
- [TEX]Q=\Delta{U}+A[/TEX](5.1)
Перший закон термодинаміки є вираженням універсального закону збереження енергії, згідно з яким енергія не може виникати нізвідкіль і зникати нікуди, однак може перетворюватися з однієї форми на іншу.
5.3 Ентальпія. Тепловий ефект реакції
Для більшості хімічних взаємодій, які найчастіше відбуваються за ізобаричних умов (Р = const), єдиним видом роботи є робота розширення:
- [TEX]A=P\cdot{\Delta{V}}[/TEX]
- де Р – зовнішній тиск,[TEX]\Delta{V}[/TEX]=V2 – V1 – змінення об’єму системи від початкового V1 до кінцевого V2. З урахуванням цього вираз першого закону термодинаміки (5.1) за умов постійного тиску набуває вигляду:
- [TEX]Q_p=\Delta{U}+P\cdot{\Delta{V}}[/TEX](5.2)
Символ Qp позначає теплоту за умови перебігу процесу при постійному тиску (Р=const).
Якщо замість[TEX]\Delta{U}[/TEX]i[TEX]\Delta{V}[/TEX]підставити відповідні значення і провести перегрупування виразу, одержуємо:
- [TEX]Q_p=U_2-U_1+P\cdot{V_2}-P\cdot{V_1}=(U_2+P\cdot{V_2})-(U_1+P\cdot{V_1})[/TEX](5.3)
Сума (U + Р·V) позначається через Н і називається ентальпією.
Ентальпія – це функція стану, що за умов постійного тиску характеризує внутрішню енергію системи та її здатність до виконання роботи.
Ентальпія залежить від кількості речовини, тому її змінення[TEX]\Delta{H}[/TEX]відносять до одного моля і вимірюють у [кДж/моль].
При підстановці Н у рівняння (5.3) одержуємо:
- [TEX]Q_p=H_2-H_1=\Delta{H}[/TEX](5.4)
Отже, в ізобаричному процесі підведена теплота дорівнює зміненню ентальпії системи.
Змінення ентальпії системи внаслідок взаємодії речовин за умов постійного тиску називається тепловий ефект хімічної реакції.
Якщо у результаті реакції ентальпія системи зменшується[TEX](H_2
Згідно з першим законом термодинаміки теплота реакції не є функцією стану, оскільки залежить від способу проведення процесу, тобто від шляху переходу системи із початкового у кінцевий стан. Однак у двох випадках теплота набуває ознак характеристичної функції. По-перше, за ізобаричних умов (Р=const,[TEX]\Delta{P}[/TEX]= 0) теплота дорівнює зміненню ентальпії:
- [TEX]Q_p=\Delta{H}[/TEX].
По-друге, коли система перебуває в ізохоричних умовах[TEX]V=const, \Delta{V}=0[/TEX], другий член у рівнянні[TEX]5.2 (Q_p=\Delta{U}+P\cdot{\Delta{V}})[/TEX]перетворюється на нуль і тоді теплота дорівнює зміненню внутрішньої енергії системи:
- [TEX]Q_V=\Delta{U}+p\Delta{V}=\Delta{U}[/TEX].
Однак хімічні реакції найчастіше відбуваються при постійному тиску, тому, крім особливо зазначених винятків, розглядають ізобаричні умови, а тепловий ефект хімічної реакції називають також ентальпія хімічної реакції[TEX]\Delta{H_T}[/TEX], де замість індексу т вказують температуру процесу.
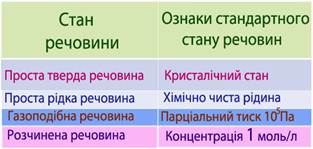
Якщо вихідні речовини і продукти реакції перебувають у стандартному стані, то тепловий ефект реакції називається стандартна ентальпія хімічної реакції[TEX]\Delta{H}[/TEX]2980.
Стандартні умови – це: Т=298К (або t=25оС) і Р=101325Па, а стандартні стани речовин наведені у табл. 5.1. Необхідно пам’ятати, що стандартні стани речовин не залежать від температури.
Для більшості хімічних реакцій змінення теплового ефекту відносно невелике, тому для його розрахунків можна знехтувати залежністю[TEX]\Delta{H}[/TEX]від температури і вважати тепловий ефект реакції постійним, тобто[TEX]\Delta{H}_Т\sim{\Delta{H}^0_{298}}[/TEX].
Таблиця 5.1 – Стандартний стан речовини
Ентальпію хімічної реакції ∆Нх.р. не слід ототожнювати з теплотою Q, незважаючи на те, що їх чисельні значення можуть співпадати. Величина Q вказує на кількість теплоти, що виділилася у навколишнє середовища (+Q) чи поглинулася з нього (–Q) під час реакції за будь-яких умов і при будь-якій довільній кількості речовини. На відміну від від теплоти Q ентальпія[TEX]\Delta{H}_T[/TEX]віднесена чітко до 1моль речовини за ізобаричних умов (Р=const). Іншою суттєвою відмінністю є знак («+» чи «–») перед величинами Q і[TEX]\Delta{H}[/TEX]. Величина ∆Нх.р. характеризує тепловий стан реакційної системи, а Q – оточуючого середовища. Екзотермічні реакції супроводжуються виділенням теплоти, отже тепловміст у системі зменшується[TEX](\Delta{H}_{продуктів}а в оточуючому середовищі, навпаки, – зростає (+Q). А при ендотермічних реакціях спостерігається зворотна залежність: за рахунок поглинання теплоти з оточуючого середовища ентальпія системи зростає[TEX](\Delta{H}_{продуктів}>\Delta{H}_{реагентів}, \Delta{H}<0)[/TEX], а теплота у навколишньому середовищі зменшується (–Q) (рис. 5.7).
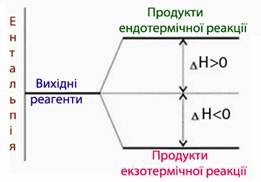
Рисунок 5.7 – Змінення ентальпії внаслідок екзо- і ендотермічної реакції
Очевидно, що для обох випадків кількість теплоти і ентальпія реакції мають протилежні знаки, тому можна вивести залежність:
- [TEX]\Delta{H}=-Q/\nu[/TEX]або[TEX]Q=-\Delta{H}\cdot{\nu}[/TEX](5.5)
- де[TEX]\nu[/TEX]– кількість речовини, моль.
На тепловий ефект реакції впливає декілька чинників, у тому числі:
- агрегатний (чи фазовий) стан вихідних речовин і продуктів реакції;
- температура. Для хімічних реакції змінення теплового ефекту в межах температур і тисків, що мають практичне значення, відносно невелике,тому для не дуже точних розрахунків можна знехтувати залежністю[TEX]\Delta{H}[/TEX]від температури і вважати тепловий ефект реакції постійним,[TEX]\Delta{H}_T[/TEX]~[TEX]\Delta{H}^0_{298}[/TEX];
- умови перебігу реакції – при сталому тиску чи при сталому об'ємі.
Тепловий ефект утворення 1моль речовини із простих сполук називається ентальпія утворення речовини, або теплотою утворення цієї речовини.
Ентальпію утворення позначають[TEX]\Delta{H_{утв}}[/TEX]або[TEX]\Delta{H_f}[/TEX], де індекс f походить від початкової букви англійського слова formation.
Ентальпію утворення простих речовин, стійких за умов 298К і 105Па, вважають такою, що дорівнює нулю:
[TEX]\Delta{H}_{утв(прост.реч-ни)}=0.[/TEX]
Якщо одна й та сама проста речовина може перебувати у різних станах, то нульове значення[TEX]\Delta{H_f}[/TEX]має фаза або модифікація, найстійкіша при 298К і 105Па, наприклад, газоподібний кисень, рідкий бром, білий фосфор, біле олово, ромбічна сірка.
Тепловий ефект реакції утворення речовин за стандартних умов називається стандартна ентальпія утворення[TEX]\Delta{H}^0_{утв.298}[/TEX], (або[TEX]\Delta{H}^0_{f,298}[/TEX]).
5.4 Основи термохімії
Термохімія – це розділ хімічної термодинаміки, що вивчає теплові ефекти хімічних реакцій та фазових перетворень.
Для термохімічних розрахунків використовують термохімічні рівняння.
Термохімічні рівняння – це рівняння реакцій, в яких вказуються агрегатні (чи фазові) стани речовин і тепловий ефект реакції, а коефіцієнтів перед формулами сполук позначають не кількість молекул, а кількість речовини.
Агрегатний стан або модифікація речовин позначається буквами: г – газоподібний, р – рідкий, т – твердий, кр – кристалічний, р-н – розчинений. Якщо агрегатні стани речовин для умов реакції очевидні, наприклад О2, N2, Al2O3 при 298 К, то їх можна не зазначати. Таким чином, будь-яку хімічну реакцію можна зобразити у вигляді відповідного термохімічного рівняння, наприклад, утворення із простих речовин водяної пари і рідкої води:
Н2 + ½ О2 = Н2О(г),[TEX]\Delta{H}^0_{298}[/TEX]= –241,8кДж,
Н2 + ½ О2 = Н2О(р),[TEX]\Delta{H}^0_{298}[/TEX]= –285,8кДж.
Термохімічні рівняння складаються так само, як і звичайні хімічні рівняння, проте в них дозволяється використання дробових коефіцієнтів, щоб теплові ефекти виражалися у кДж/моль переважно для однієї з вихідних або кінцевих речовин. Наприклад, хімічному рівнянню реакції
2С6Н6 + 15О2[TEX]\rightarrow[/TEX]12CO2 + 6H2O
відповідає термохімічне рівняння, в якому відображається згоряння 1моль бензену, вказуються агрегатні стани речовин, використовуються дробові коефіцієнти та наводиться тепловий ефект:
C6H6(р) +7,5O2[TEX]\rightarrow[/TEX]6CO2+3H2O(р),[TEX]\Delta{H}^0_{298}[/TEX]= –3267,7кДж/моль.
При термохімічних розрахунках слід пам’ятати перший закон термохімії, відомий ще під назвою закон Лавуазьє-Лапласа (1784 р.), який спочатку був сформульований так: «При розкладанні складної сполуки на прості поглинається (чи виділяється) стільки теплоти, скільки її виділяється (чи поглинається) при утворенні такої ж кількості складної сполуки із простих».
Сучасне формулювання перший закон термохімії:
Ентальпія утворення складної сполуки чисельно дорівнює ентальпії її розкладання, взятій з протилежним знаком.
[TEX]\Delta{H}|^0_{утв.(складн.реч-ни)}=-\Delta{H}^0_{розкл.(складн.реч-ни)}[/TEX].
Наприклад,[TEX]\Delta{H}^0_{утв.}(Н_2О_{(г)})=-\Delta{H}^0_{розкл.}(Н_2О_{(г)}).[/TEX]
Незалежність теплоти хімічної реакції від шляху процесу за ізобарно-ізотермічних умов (Р,Т = const) була встановлена на основі експериментальних досліджень і має назву закон Гесса, або другого закону термохімії:
тепловий ефект хімічної реакції за умов сталого тиску і сталої температури не залежить від шляху її перебігу, а залежить лише від природи і фізичного стану вихідних речовин і продуктів реакції.
Якщо уявити, що від реагентів із початкового стану (рис. 5.8) можна перейти до продуктів реакції у кінцевий стан декількома шляхами через різні проміжні стадії, кожна з яких має власний тепловий ΔH1, ΔH2, ... ΔH8, то відповідно до закону Гесса, тепловий ефект ΔH1 прямого переходу від початкового стану в кінцевий пов’язаний з тепловими ефектами інших переходів рівністю:
ΔH1 = ΔH2 + ΔH3 + ΔH4 + ΔH5 + ΔH6 = ΔH7+ ΔH8 .
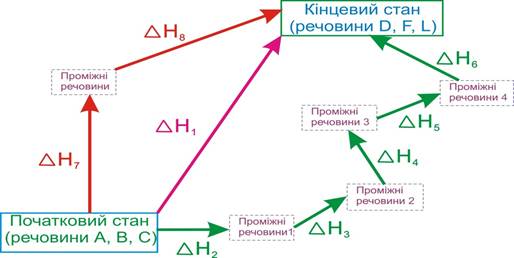
Рисунок 5.8 – Ілюстрація до закону Гесса для реакції А + B + C = D + F + L
Аналогічна залежність справедлива і для фазових перетворень, наприклад (рис. 5.9), від твердого стану речовина може перейти у газоподібний внаслідок або безпосередньої сублімації, або послідовного протікання процесів плавлення і випаровування речовини. При цьому теплові ефекти перетворень співвідносяться згідно із законом Гесса:
ΔHсублімації = ΔHплавлення + ΔHвипаровування.

Рисунок 5.9 – Теплові ефекти фазових переходів
Закон Гесса використовують для багатьох хімічних розрахунків, у тому числі для обчислення теплових ефектів реакцій, які технічно складно чи зовсім неможливо встановити експериментально. Наприклад, перейти від графіту і О2 до карбон (ІV) оксиду можна двома способами (рис.5.10): або через проміжну стадію утворення СО і подальшого його доокиснення за рівняннями
С(графіт) + ½ О2(г) = СО(г),[TEX]\Delta{H}_1[/TEX],
СО(г) + ½ О2(г) = СО2(г),[TEX]\Delta{H}_2[/TEX]= –283,1кДж,
або при безпосередній взаємодії простих речовин
С(графіт) + О2(г) = СО2(г),[TEX]\Delta{H}_3[/TEX]= –393,6кДж.
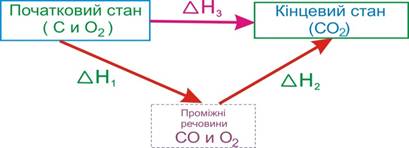
Рисунок 5.10 – Теплові ефекти реакцій утворення СО2 двома способами
Згідно з законом Гесса тепловий ефект утворення СО2 з простих речовин дорівнює сумарному тепловому ефекту утворення СО2 через проміжну стадію:
[TEX]\Delta{H}_3=\Delta{H}_1+\Delta{H}_2[/TEX].
У розглянутій схемі можна експериментально визначити теплові ефекти[TEX]\Delta{H}_2[/TEX]і[TEX]\Delta{H}_3[/TEX], а тепловий ефект[TEX]\Delta{H}_1[/TEX], виміряти який надзвичайно складно, обчислюють:
[TEX]\Delta{H}_1=\Delta{H}_3-\Delta{H}_2=-393,6-(-283,1)=-110,5[/TEX]кДж.
Із закону Гесса випливають важливі наслідки.
тепловий ефект зворотної реакції дорівнює тепловому ефекту прямої реакції, взятому з протилежним знаком (рис.5.11):
[TEX]\Delta{H}_{пр}=-\Delta{H}_{зворотн}[/TEX].
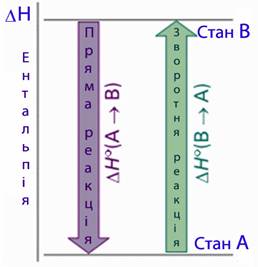
Рисунок 5.11 – Перший наслідок закону Гесса
Отже, закон Гесса доводить, що яким би шляхом не перебігала реакція, її тепловий ефект буде однаковим, якщо при цьому не змінюється кінцевий і вихідний стани системи.
ентальпія хімічної реакції дорівнює сумі ентальпій утворення продуктів реакції за винятком суми ентальпій утворення вихідних речовин з урахуванням відповідних стехіометричних коефіцієнтів.
Другий наслідок закону Гесса дає можливість розрахувати ентальпію хімічної реакції. Для реакції загального вигляду
bВ + dD = lL + mM,
де символами B, D, L, M зашифровані формули речовин, а буквами b, d, l, m – коефіцієнти перед ними, тепловий ефект обчислюється на основі другого наслідку закону Гесса:
- [TEX]\Delta{Н}^0_{х.р}=l\Delta{H}^0_{f,L}+m\Delta{H}^0_{f,M}-b\Delta{H}^0_{f,B}-d\Delta{H}^0_{f,D}[/TEX].(5.6)
На основі закону Гесса можна розрахувати ентальпія утворення будь-якої речовини, якщо відомі ентальпії утворення усіх інших речовин і ентальпія хімічної реакції, наприклад, ентальпія утворення складної сполуки М обчислюється так:
- [TEX]\Delta{H}^0_{f,M}=(\Delta{H}^0_{x.p.}-l\Delta{H}^0_{f,L}+d\Delta{H}^0_{f,D}+b\Delta{H}^0_{f,B})/m[/TEX].
Оскільки ентальпія хімічної реакції є наслідком руйнування одних хімічних зв’язків і утворення інших, то за відомими значеннями енергії хімічних зв’язків можна обчислити ентальпію хімічної реакції або за відомою ентальпією – енергію зв’язку.
Перебіг хімічних реакцій дуже часто супроводжується фазовими чи поліморфними перетвореннями, які теж характеризуються власними енергетичними ефектами. Процеси переходу твердого тіла у рідину (плавлення) і газ (сублімація), рідини у газ (пароутворення), кристалічного стану в аморфний, менш стійкої модифікації у більш стійку є ендотермічними. Зворотні процеси – кристалізації, конденсації, переходу аморфного стану до кристалічного – протікають екзотермічно. Теплові ефекти фазових та поліморфних перетворень, як правило, суттєво менші, ніж теплові ефекти хімічних реакцій.
Застосування закону Гесса надзвичайно поширює можливості термохімії, дозволяючи виконувати точні розрахунки ентальпій утворення цілого ряду речовин, дослідні дані для яких важко було одержати з технічних причин. На основі закону Гесса обчислюються термодинамічні функції, що використовуються у безлічі термохімічних і термодинамічних розрахунків.
5.5 Ентропія. Другий закон термодинаміки
При вивченні хімічних процесів надзвичайно важливо оцінити принципову можливість чи неможливість їх перебігу, а також напрямок і межі самочинного перебігу реакцій за даних умов.
Самочинні процеси – це такі процеси, що перебігають без підведення енергії ззовні.
Як приклади самочинних фізичних процесів можна навести передавання теплоти від нагрітого тіла холодному (рис. 5.12 а), здатність молекул газу займати весь об’єм посудини (рис. 5.12 б), а приклади хімічних реакцій – утворення іржі на металах, розчинення солі у воді тощо.
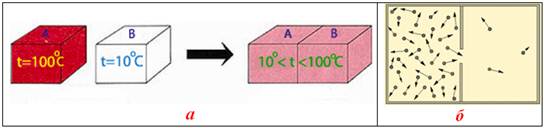
Рисунок 5.12 – Самочинні фізичні процеси
Рушійними силами самочинного перебігу процесів є два фактори:
- прагнення системи до мінімуму енергії;
- прагнення до досягнення найбільш імовірного за даних умов стану.
Перший фактор, який одержав назву ентальпійного, виявляється у зміненні ентальпії. Це пояснюється так: під час будь-якої хімічної реакції відбувається розрив зв'язків у молекулах вихідних реагентів, який потребує витрати енергії, і одночасно – утворення нових зв'язків у молекулах продуктів реакції, яке супроводжується виділенням енергії. Зрозуміло, що з більшою імовірністю самочинно процес буде перебігати у тому випадку, коли витрата енергії на руйнування зв'язків компенсується виграшем енергії при їх утворенні. Іншими словами, якщо енергії виділяється більше, ніж витрачається, тобто[TEX]\Delta{H}_{x.p.}<0[/TEX].
І дійсно, безліч хімічних реакцій протікає самочинно з виділенням енергії і зниженням тепловмісту системи (екзотермічні процеси,[TEX]\Delta{H}<0[/TEX]). Виходячи із спостережень, Бертло (1867р.) сформулював свою гіпотезу, відому нині як
Принцип самочинного перебігу хімічних реакцій:
самочинно перебігають лише ті процеси, що супроводжуються виділенням теплоти.
Однак, досвід свідчить, що умова[TEX]\Delta{H}>0[/TEX]не може бути вичерпним критерієм, оскільки існують і самочинні ендотермічні процеси, для яких[TEX]\Delta{H}<0[/TEX](наприклад, розчинення NH4NO3 i KCl у воді), і навпаки, деякі екзотермічні реакції за стандартних умов не здійснюються, наприклад, синтез амоніаку NH3.
У такому разі що ж може служити рушійною силою ендотермічних реакцій і тих процесів, що не супроводжуються тепловими ефектами? Уявимо простий дослід (рис. 5.13). Якщо через отвір сполучити дві посудини – з бурим газом NO2 і безбарвним азотом N2, що перебувають за однакових умов (температури, тиску), то через деякий час забарвлення в обох посудинах вирівнюється.
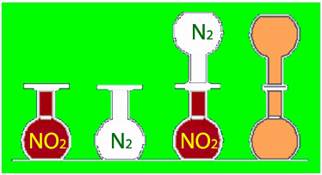
Рисунок 5.13 – Самочинне змішування бурого газу NO2 і безбарвного азоту N2
Оскільки між газами NO2 і N2 не відбувається взаємодії (зв’язки не руйнуються і не утворюються), то тепловий ефект відсутній,[TEX]\Delta{H}=0[/TEX]. З цього випливає, що ентальпійний фактор не може вважатися рушійною силою такого процесу.
Отже, крім ентальпійного фактору, існує й інша рушійна сила самочинного перебігу процесів – це здатність частинок (молекул, йонів, атомів) до хаотичного руху, внаслідок якого система набуває найбільш імовірного стану і переходить із більш упорядкованого стану (індивідуальні гази) у менш упорядкований (суміш газів). Імовірність зворотного переходу в упорядкований стан, коли суміш газів самочинно розподіляється на індивідуальні гази, практично дорівнює нулю, тому що такий процес потребує затрати енергії ззовні.
Описаний дослід є спрощеною ілюстрацією до загального закону природи, згідно з яким статистичні системи (тобто такі, що складаються з величезної кількості частинок) завжди прагнуть досягти найбільш невпорядкованого стану.
Для оцінки ступеня невпорядкованості системи введено спеціальну термодинамічну функцію – ентропію S.
Ентропія – це термодинамічна функція, яка є мірою невпорядкованості і характеризує відносну імовірність стану системи.
Основною властивістю ентропії є її збільшення[TEX](\Delta{S}>0)[/TEX]) у будь-якій замкнутій системі, що змінює свій стан у напрямку рівноваги, якій притаманні максимальна невпорядкованість частинок і найбільші значення ентропії[TEX](\Delta{S}_{рівноваги}=0)[/TEX].
Розглянемо ще один уявний експеримент. Нехай є ізольована система, розділена перетинкою на дві рівні частини, в одній з яких міститься чотири молекули (рис.5.14).
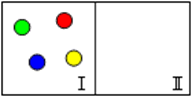
Рисунок 5.14 – Модель ізольованої системи, що розділена на дві рівні частини і містить чотири молекули
Якщо в деякий момент прибрати перетинку, то завдяки хаотичному руху молекул можуть виникати різні варіанти їх розподілу по обох частинах посудини. Кожний варіант – це окремий мікростан системи (табл. 5.2).
Таблиця 5.2 – Можливі варіанту розподілу чотирьох молекул по двох половинах посудини і термодинамічна імовірність стану системи
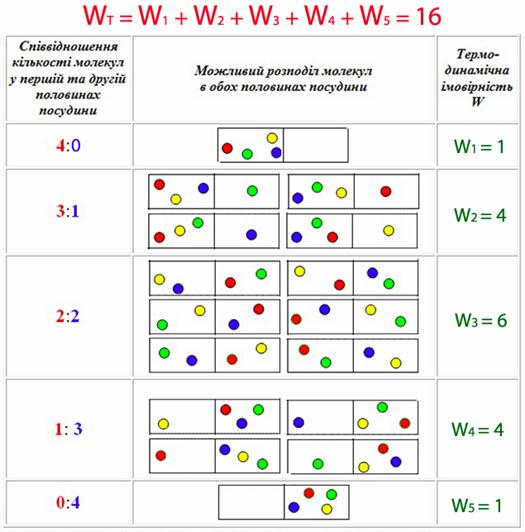
Рівномірний розподіл частинок між окремими ділянками об’єму здійснюється лише як середній за часом. У кожний даний момент внаслідок хаотичного руху спостерігається тимчасове збільшення концентрації частинок на одних ділянках об’єму і зменшення її на інших – флуктуації густини. Макроскопічний стан усієї системи в цілому може існувати при різному розподілі частинок (тобто при різних мікроскопічних станах) і буде характеризуватися мікростанами складових частин, які описуються миттєвими координатами частинок та швидкостями різних видів руху в різних напрямках.
Кількість мікростанів, з яких складається даний макроскопічний стан системи, називається термодинамічна імовірність W.
На практиці мають справу з величезним числом частинок у системі, що позначається на великих значеннях термодинамічної імовірності, тому користуються не абсолютною величиною W, а її логарифмом lnW, який пов’язаний з ентропією залежністю:
- [TEX]S=k\cdot{\ln{W}}[/TEX],(5.7)
де k – стала Больцмана (k = 1,38·10–23Дж/К), яка в свою чергу пов’язана з універсальною молярною сталою R і числом Авогадро NA (k=R/NA). З урахуванням цього зв’язку для одного моля речовини ([TEX]\nu=1моль[/TEX]) рівняння (5.7) набуває вигляду:
- [TEX]S=R\cdot{\ln{W}}[/TEX].(5.8)
Із (5.8) видно, що ентропія, як і молярна стала R (R = 8,314 Дж/моль·К), вимірюється у [Дж/моль×К].
Ентропія речовини у стандартному стані називається стандартна ентропія S0298.
На відміну від інших термодинамічних функцій можна визначити не тільки змінення ентропії[TEX]\Delta{S}[/TEX], але й її абсолютні значення. Це випливає із сформульованого Планком (1911 р.) постулату, більше відомого як
при абсолютному нулі (0 K) ентропія ідеального кристалу дорівнює нулю.
У міру віддалення від абсолютного нуля при підвищенні температури зростає енергія та швидкість руху частинок, збільшується кількість мікростанів, тому підвищується термодинамічна імовірність і, відповідно, ентропія (рис. 5.15). При переході речовини із одного агрегатного стану в інший різко змінюється невпорядкованість системи, тому ентропія змінюється стрибкоподібно[TEX](\Delta{S}_{плавл},\Delta{S}_{кип})[/TEX]).
Значення ентропії складним чином відображує всю сукупність властивостей сполуки. На величину ентропії речовин впливають різні фактори:
- агрегатний стан. Ентропія зростає при переході із твердого стану в рідкий, і особливо, у газоподібний;
- молекулярна маса. Ентропія збільшується із зростанням молекулярної маси у ряді близьких за властивостями речовин, наприклад, для атомарного і молекулярного кисню та озону: S0298(0)=161Дж/моль·К; S0298(O2)=205Дж/моль·К; S0298(O3)=239Дж/моль·К;
- будова твердого тіла. Ентропія речовини з більш упорядкованою кристалічною граткою нижча за ентропію тієї самої речовини з менш упорядкованою кристалічною структурою, а ентропія речовини в аморфному стані вища, ніж ентропія цієї речовини у кристалічному стані;
- ізотопний склад, наприклад, для важкої і звичайної води: S0298(D2O) > S0298(H2O);
- будова молекул, наприклад, ентропія ізомерів нормальної будови менше, ніж розгалужених: S0298(ізобутану) > S0298(н-бутану).
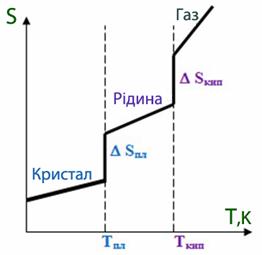
Рисунок 5.15 – Характер змінення ентропії[TEX](\Delta{S})[/TEX]речовини під час фазових перетворень при підвищенні температури
Ентропія S – це термодинамічна функція, тому, як і для будь-якої іншої термодинамічної функції, її змінення[TEX](\Delta{S})[/TEX]не залежить від шляху переходу системи з початкового у кінцевий стан, отже
Змінення ентропії[TEX]\Delta{S}[/TEX]під час перебігу хімічної реакції дорівнює сумі ентропій продуктів реакції за винятком суми ентропій вихідних речовин з урахуванням стехіометричних коефіцієнтів:
[TEX]\Delta{S}_{х.р}=\sum{S}_{f,продуктів}-\sum{S}_{f,вих.речовин}[/TEX].
Для реакції загального вигляду
bB + dD = lL + mM,
де символами B, D, L, M зашифровані формули речовин, а буквами b, d, l, m – коефіцієнти перед ними, змінення ентропії системи (або просто ентропії хімічної реакції) дорівнює:
- [TEX]\Delta{S}=(lS_{f,L}+mS_{f,M})-(bS_{f,B}+dS_{f,D})[/TEX].(5.9)
При якісному оцінюванні змінення ентропії реакції корисно пам'ятати правило:
Оскільки ентропія речовини у газоподібному стані істотно вища, ніж у рідкому і твердому станах, то ентропія реакції додатна[TEX](\Delta{S}>0)[/TEX]), якщо внаслідок процесу збільшується кількість молей газу.
З поняттям ентропія пов’язаний другий закон термодинаміки, що має декілька формулювань. Для систем, у яких відсутній енергообмін і масообмін з навколишнім середовищем, він формулюються так:
Другий закон термодинаміки:
в ізольованих системах самочинно перебігають тільки ті процеси, що супроводжуються зростанням ентропії.
Другий закон термодинаміки не є абсолютним законом природи, подібно до першого закону, а має статистичний характер, тобто поширюється лише на системи, які складаються з великої кількості частинок.
Хімічні реакції, які у більшості випадків відбуваються в неізольованих системах, не підлягають другому закону термодинаміки. Частина процесів протікає зі зменшенням ентропії і супроводжується зміненням внутрішньої енергії (тепловий ефект) завдяки тому, що система обмінюється енергією з навколишнім середовищем. Коли хімічні реакції відбуваються із зменшенням ентропії, то зростає ентропія навколишнього середовища. Наприклад, хімічні реакції в організмі будь-якої живої істоти супроводжуються зменшенням ентропії, тому що збільшується впорядкованість системи. Але організм одержує енергію з навколишнього середовища (повітря, харчі), внаслідок чого зростає ентропія саме навколишнього середовища.
Під час перебігу хімічних реакцій система обмінюється енергією з навколишнім середовищем, тобто вона не є ізольованою. При цьому, як правило, змінюються і ентропія і ентальпія.
Отже, в хімічних реакціях виявляються дві тенденції. Перша пов’язана з прагненням системи до утворення міцних зв’язків між частинками і виникнення більш складних сполук, що супроводжується зниженням внутрішньої енергії системи. За ізобарно-ізотермічних умов це пов’язано з характеристикою, що називається ентальпійний фактор і виражається через[TEX]\Delta{H}[/TEX](кДж/моль). Друга тенденція виявляється у прагненні до роз'єднання частинок, до безладу і зростання ентропії. Ця ентропійний фактор, який кількісно виражається добутком абсолютної температури на ентропію[TEX]T\cdot{\Delta{S}}[/TEX](кДж/моль).
5.6 Напрям перебігу хімічних реакцій. Енергія Гіббса
Ентальпійний і ентропійний фактори, що відображають дві протилежні тенденції, не можуть бути вичерпним критерієм самочинного протікання процесів. Для ізобарно-ізотермічних процесів їх об’єднує функція, яка називається енергією Гіббса[TEX](\Delta{G})[/TEX]і дорівнює:
- [TEX]\Delta{G}=\Delta{H}-T\cdot{\Delta{S}}[/TEX].(5.10)
Енергія Гіббса вимірюється в [кДж/моль].
Рівняння (5.10) можна записати у вигляді:
- [TEX]\Delta{Н}=\Delta{G}+T\cdot{\Delta{S}}[/TEX].(5.11)
Із (5.11) видно, що ентальпія хімічної реакції містить дві частини. Перший член рівняння [TEX](\Delta{G})[/TEX]дорівнює максимальній роботі Арmax, яку може виконати система при рівноважному проведенні процесу за ізобарно-ізотермічних умов, тобто
Енергія Гіббса – це частина енергетичного ефекту хімічної реакції, яку можна перетворити в роботу:
- [TEX]-\Delta{G}=A^{max}_{p}[/TEX].(5.12)
Знак "мінус" позначає, що система здатна виконати роботу над навколишнім середовищем тільки за рахунок зменшення енергії Гіббса внаслідок реакції. Оскільки енергію Гіббса можна перетворити у роботу, то її іноді називають вільною енергією.
Другий член правої частини рівняння (5.11) – ентропійний фактор – являє собою частину енергетичного ефекту, яку неможливо перетворити у роботу. Ця частина розсіюється у навколишнє середовище у вигляді теплоти, тому ентропійний фактор[TEX]T\Delta{S}[/TEX]називають зв’язаною енергією.
Енергія Гіббса[TEX]\Delta{G}[/TEX]є критерій самочинного перебігу хімічної реакції, тому знак перед чисельним значенням[TEX]\Delta{G}[/TEX](«+» чи «–») дозволяє зробити висновок про принципову можливість чи неможливість самочинного протікання реакції.
- Зменшення енергії Гіббса[TEX](\Delta{G}<0)[/TEX]свідчить про те, що самочинний перебіг реакції у прямому напрямку за даних умов є принципово можливим (рис.5.16).
- Збільшення енергії Гіббса[TEX](\Delta{G}>0)[/TEX]є умовою неможливості самочинного протікання прямої реакції за даних умов.
- Якщо енергія Гіббса не змінюється[TEX](\Delta{G}=0)[/TEX], то можливе самочинне протікання реакції як у прямому, так і у зворотному напрямку, тобто система перебуває у стані рівноваги.
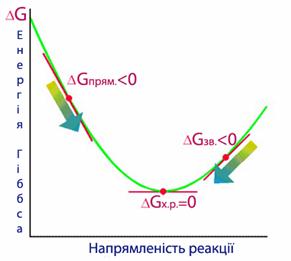
Рисунок 5.16 – Енергія Гіббса – критерій можливості самочинного перебігу реакцій: пряма реакція проходить самочинно якщо[TEX]\Delta{G}<0[/TEX], і навпаки, при цьому неможливо самочинне протікання зворотної реакції, для якої[TEX]\Delta{G}>0[/TEX]. За умови[TEX]\Delta{G}=0[/TEX]у реакційній системі встановлюється рівновага.
В стані рівноваги енергія Гіббса не змінюється, тому[TEX]\Delta{G}=0[/TEX]. З іншого боку енергія Гіббса визначається рівнянням (5.10:[TEX]\Delta{G}=\Delta{H}-T\cdot{\Delta{S}}[/TEX]). Порівнюючи обидва рівняння, неважко визначити температуру Трівн, при якій наступає стан рівноваги
[TEX]\Delta{G}=\Delta{H}-Т_{рівн}\cdot{\Delta{S}}=0[/TEX],
тому[TEX]\Delta{H}=T_{рівн}\cdot{\Delta{S}}[/TEX],
звідки температура, при якій у системі наступає рівновага:
- [TEX]T_{рівн}=\Delta{H}/\Delta{S}[/TEX].(5.13)
На основі рівняння (5.13) можна обчислити температуру, вище якої відбувається змінення знаку енергії Гіббса на протилежний і, як наслідок, змінення напрямку протікання реакції з прямого на зворотний.
Знак енергії Гіббса[TEX]\Delta{G}[/TEX]дозволяє визначити з достатньою вірогідністю напрямок самочинного перебігу будь-якої реакції за відомими значеннями[TEX]\Delta{H}[/TEX]і[TEX]\Delta{S}[/TEX]при певній температурі. Розглянемо декілька типових випадків (табл.5.3).
- Екзотермічні реакції[TEX](\Delta{H}<0)[/TEX]завжди перебігають самочинно у прямому напрямку, якщо внаслідок реакції збільшується кількість молів газоподібних речовин і, відповідно, зростає ентропія[TEX]\Delta{S}>0[/TEX]. При цьому енергія Гіббса набуває від’ємних значень,[TEX]\Delta{G}=\Delta{H}-T\cdot{\Delta{S}}<0[/TEX].
- Якщо у результаті екзотермічної реакції[TEX](\Delta{H}<0)[/TEX]ентропія зменшується[TEX](\Delta{S}<0)[/TEX], то за умов низьких температур ентальпійний фактор переважає над ентропійним[TEX](|\Delta{H}|>|T\Delta{S}|)[/TEX]і[TEX]\Delta{G}<0[/TEX], тобто реакція самочинно перебігає у прямому напрямку. Однак після досягнення рівноважної температури Трівн співвідношення ентальпійного та ентропійного факторів змінюється[TEX](|\Delta{H}|<<|T\Delta{S}|)[/TEX], а енергія Гіббса набуває додатних значень[TEX](\Delta{G}>0)[/TEX], тому стає неможливим самочинний перебіг прямої реакції, але перебігає зворотна реакція.
- Ендотермічна реакція[TEX](\Delta{H}<0)[/TEX], у результаті якої зменшується ентропія[TEX](\Delta{S}<0)[/TEX], не може протікати самочинно у прямому напрямку за будь-яких температур, оскільки завжди[TEX]\Delta{G}>0[/TEX].
- Якщо внаслідок ендотермічної реакції[TEX](\Delta{H}>0)[/TEX]збільшується ентропія системи[TEX](\Delta{S}>0)[/TEX], то за низьких температур, коли[TEX](|\Delta{H}|>|T\Delta{S}|)[/TEX], самочинно пряма реакція відбуватися не може[TEX](\Delta{G}>0)[/TEX], а за високих температур (Т>Трівн) пряма реакція перебігає самочинно.
Таблиця 5.3 – Вплив температури на напрямок хімічних реакцій
| [TEX]\Delta{H}[/TEX] | [TEX]\Delta{S}[/TEX] | [TEX]\Delta{G}[/TEX] | Напрям самочинного перебігу реакції | Приклад реакції |
|
[TEX]\Delta{H}<0[/TEX] |
[TEX]\Delta{S}>0[/TEX] | [TEX]\Delta{G}<0[/TEX] |
Перебіг прямої реакції можливий при будь-якій температурі |
[TEX]C_{графит}+\frac{1}{2}O_2\Rightarrow{CO}[/TEX] |
| [TEX]\Delta{H}<0[/TEX] | [TEX]\Delta{S}<0[/TEX] | [TEX]\Delta{G}<0[/TEX]при Т<Трівн [TEX]\Delta{G}>0[/TEX]при Т>Трівн |
При низьких температурах можливий перебіг прямої реакції, а при високих – зворотної |
[TEX]CaO+CO_2\Leftrightarrow{CaCo_3}[/TEX] |
| [TEX]\Delta{H}>0[/TEX] | [TEX]\Delta{S}<0[/TEX] | [TEX]\Delta{G}>0[/TEX] |
Перебіг прямої реакції нездійснений при будь-якій температурі |
[TEX]CO\leftarrow{C_{графит}+\frac{1}{2}O_2}[/TEX] |
| [TEX]\Delta{H}>0[/TEX] | [TEX]\Delta{S}>0[/TEX] | [TEX]\Delta{G}>0[/TEX]при Т<Трівн [TEX]\Delta{G}<0[/TEX]при Т>Трівн |
При високих температурах можливий перебіг прямої реакції, а при низьких – зворотної |
[TEX]CH_4+2H_2O_{(г)}\Leftrightarrow{CO_2+4H_2}[/TEX] |
Змінення енергії Гіббса системи при утворенні одного моля сполуки із простих речовин, стійких при 298К, називається енергія Гіббса утворення цієї сполуки[TEX]\Delta{G}_f[/TEX].
Енергія Гіббса утворення простих речовин вважається такою, що дорівнює нулю:[TEX]\Delta{G}_{f(прост.реч.)}=0[/TEX].
Наприклад, енергія Гіббса утворення амоніаку[TEX]\Delta{G}_f(NH_3_{(г)})[/TEX]дорівнює енергії Гіббса реакції ½ N2 + 3/2 H2 = NH3.
Якщо речовина і вихідні прості сполуки, з яких вона утворена, перебувають у стандартних станах (див. табл. 5.1), то енергія Гіббса утворення називається стандартна енергія Гіббса даної речовини[TEX]\Delta{G}^0_f[/TEX].
Як і будь-яка термодинамічна функція, енергія Гіббса є функцією стану, тому її значення не залежать від шляху протікання процесу, а визначається лише початковим і кінцевим станами системи.
Енергія Гіббса хімічної реакції обчислюється як сума енергій Гіббса утворення продуктів реакції за винятком суми енергій Гіббса утворення вихідних речовин з урахуванням стехіометричних коефіцієнтів:
[TEX]\Delta{G}_{х.р}=\sum{G}_{f,прод}-\sum{G}_{f,вих.реч-н}[/TEX].
Енергія Гіббса хімічної реакції загального вигляду
- [TEX]dD+bB=lL+mM[/TEX],
де символами B, D, L, M зашифровані формули речовин, а буквами b, d, l, m – коефіцієнти перед ними, змінення енергії Гіббса розраховується згідно з формулою:
- [TEX]\Delta{G}=l\Delta{G}_{f,L}+mDG_{f,M}-d\Delta{G}_{f,D}-b\Delta{G}_{f,B}[/TEX].(5.14)
Якщо вихідні речовини і продукти реакції перебувають у стандартних станах, то енергія Гіббса називається стандартна енергія Гіббса хімічної реакції[TEX]\Delta{G}^0_{х.р.}[/TEX]і є критерієм самочинного протікання реакції за стандартних умов для вихідних речовин і продуктів реакції.
5.7 Приклади розв'язання типових задач
Приклад 5.1. Визначити кількість теплоти, що поглинається при утворенні 11,2 л NO за стандартних умов, якщо тепловий ефект реакції[TEX]\Delta{H}^0_{х.р.}=+180,8[/TEX]кДж.
Розв’язок. Складаємо термохімічне рівняння реакції
N2(г) + O2(г)[TEX]\Rightarrow[/TEX]2NO(г),[TEX]\Delta{H}^0_{х.р.}= +180,8[/TEX]кДж.
Обчислюємо кількість речовини NO:
ν(NO) =V/VM = 11,2л / 22,4л/моль = 0,5 моль.
З урахуванням рівняння (5.5), яке зв'язує довільну кількість теплоти з тепловим ефектом реакції, маємо:
[TEX]Q =-\Delta{H}\cdot{\nu}[/TEX]= –180,0кДж/моль·0,5моль = –45,2кДж.
Приклад 5.2. Обчислити ентальпію конденсації водяної пари. Чи виділяється при цьому теплота?
Розв’язок. Термохімічне рівняння конденсації водяної пари має вигляд:
Н2О(г) = Н2О(р),[TEX]\Delta{H}^0_{298}[/TEX]–?
Скористаємося наслідком закону Гесса і довідниковими даними щодо ентальпій утворення відповідних речовин:
[TEX]\Delta{H}^0_{298}=\Delta{H}^0_f(Н_2О_{(р)}-\Delta{H}^0_f (Н_2О_{(г)})=-241,8-(-285,8)=+44[/TEX]кДж.
У результаті конденсації водяної пари ентальпія зростає[TEX](\Delta{H}^0_{298}>0)[/TEX], тобто система поглинає теплоту, тому процес конденсації є ендотермічним.
Приклад 5.3. Скласти термохімічне рівняння реакції згоряння етанолу, якщо відомо, що при спалюванні 4,6г С2Н5ОН виділяється 136,7 кДж теплоти.
Розв’язок. Для складання термохімічного рівняння необхідно обчислити тепловий ефект реакції у розрахунку на 1моль С2Н5ОН. Знайдемо кількість речовини етанолу:
ν(С2Н5ОН) = m/M =4,6 / 46 = 0,1моль.
Відповідно до умови задачі кількість теплоти Q =136,7кДж, тоді тепловий ефект реакції
[TEX]\Delta{H}=-Q/\nu=-137/0,1=-1370[/TEX]кДж/моль.
Термохімічне рівняння реакції має вигляд:
- [TEX]C_2H_5OH_{(р)}+3O_{2(г)}=2CO_{2(г)}+3H_2O_{(р)}; \Delta{H}^0_{х.р}=-1370[/TEX]кДж.
Приклад 5.4. Обчислити тепловий ефект реакції між сульфур(ІV) оксидом і сірководнем за даними ентальпій утворення речовин (кДж/моль):[TEX]\Delta{H}^0_f(SO_2)=-296,9; \Delta{H}^0_f(H_2S)=-20,15; \Delta{H}^0_f(H_2O)_{(p)}=-285,84.[/TEX]Чи належить ця реакція до екзотермічних?
Розв’язок. Для заданої реакції
SO2(г) + 2H2S(г) = 3S(т) + 2H2O(р)
тепловий ефект обчислюється за наслідком закону Гесса:
- [TEX]\Delta{H}^0_{298}=3\Delta{H}^0_f(S)+2\Delta{H}^0_f(H_2O)-\Delta{H}^0_f(SO_2)-2\Delta{H}^0_f(H_2S)=3\cdot{0}+2(-285,84)-(-296,9)-2(-20,15)=-234,4[/TEX]кДж.
Оскільки в результаті реакції ентальпія системи зменшується[TEX](\Delta{H}^0_{298}<0)[/TEX], то відбувається виділення теплоти, отже, реакція екзотермічна.
Приклад 5.5. Тепловий ефект реакції згоряння 1 моль рідкого бензену з утворенням карбон диоксиду і водяної пари дорівнює –3135,58кДж. Написати термохімічне рівняння і обчислити ентальпію утворення С6Н6(р).
Розв’язок. Термохімічне рівняння має вигляд:
[TEX]C_6H_6_{(р)}+7,5О_2_{(г)}=6CO_{2(г)}+3H_2O_{(г)}; \Delta{H}^0_{298}=-3135,58[/TEX]кДж.
Згідно із законом Гесса тепловий ефект цієї реакції:
[TEX]\Delta{H}^0_{298}=6\Delta{H}^0_f(СО_2)+3\Delta{H}^0_f(H_2O_{(г)})-\Delta{H}^0_f(C_6H_{6(р)})-7,5\Delta{H}^0_f(O_2)[/TEX],
звідки ентальпія утворення рідкого бензену:
- [TEX]\Delta{H}^0_f(C_6H_{6(p)})=6\Delta{H}^0_f(CO_2)+3\Delta{H}^0_f(H_2O_{(г)})-7,5\Delta{H}^0_f(O_2)-\Delta{H}^0_{298}[/TEX].
Користуючись довідковими даними щодо ентальпій утворення відповідних речовин, одержимо:
- [TEX]\Delta{H}^0_f(C_6H_{6(p)})=6(-393,51)+3(-241,83)-7,5\cdot{0}-(-3135,58)=+49,03[/TEX]кДж/моль.
Приклад 5.6. Визначити інтервал температур, за яких реакція СаО + СО2 = СаСО3 протікає у прямому напрямку, а за яких – у зворотному.
Розв’язок. На основі довідкових даних щодо ентальпій та ентропій утворення відповідних речовин обчислимо змінення ентропії і тепловий ефект реакції
СаО(т) + СО2(г) ↔ СаСО3(т).
Оскільки ентропія є функцією стану, її змінення внаслідок хімічної реакції дорівнює різниці ентропій утворення продуктів реакції та вихідних речовин:
- [TEX]\Delta{S}^0_{298}=S^0_{298}(CaCO_3)-S^0_{298}(Ca)-S^0_{298}(CO_2)=92,88-39,7-213,68=-160,5[/TEX]Дж/мольK[TEX]=-0,1605[/TEX]кДж/моль К.
Згідно із законом Гесса розрахуємо тепловий ефект:
- [TEX]\Delta{Н}^0_{298}=\Delta{H}^0_{f}(CaCO_3)-\Delta{H}^0_f(Ca)-\Delta{H}^0_f(CO_2)=-1207,1-(-635,5)-(-393,51)=-178,1[/TEX]кДж/моль.
Для заданої реакційної системи (СаО(т)+СО2(г)↔СаСО3(т)) пряма реакція – екзотермічна[TEX](\Delta{H}^0_{298}<0)[/TEX], вона супроводжується зменшенням ентропії[TEX](\Delta{S}^0_{298}<0)[/TEX]. Спираючись на дані табл.5.3, можна зробити попередній висновок, що за низьких температур (Т<Трівн) самочинно буде протікати саме пряма реакція, а за високих (Т>Трівн) – зворотна. Однак точнішу відповідь одержимо на підставі розрахунків енергії Гіббса. За стандартних умов (Т=298 К) змінення енергії Гіббса залежить від співвідношення ентальпійного і ентропійного фокторів:
- [TEX]\Delta{G}^0_{298}=\Delta{H}-T\Delta{S}=-178,1-298(-0,1605)=-130,3[/TEX]кДж/моль К.
Як показують розрахунки, внаслідок реакції енергія Гіббса зменшується[TEX](\Delta{G}^0_{298}<0)[/TEX], що доводить можливість самочинного перебігу реакції.
Для визначення рівноважної температури, при якій відбувається змінювання знака енергії Гіббса, скористаємося умовою[TEX]T_{рівн}=\Delta{H}\Delta{S}[/TEX], і будемо вважати, що[TEX]\Delta{H}^0_{298}[/TEX]і[TEX]\Delta{S}^0_{298}[/TEX]мало змінюються при підвищенні температури. Тоді
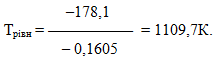
Отже, при температурах, нижче 1109,7К самочинно буде протікати пряма реакція, а вище 1109,7К – зворотнa.
Приклад 5.7. На основі довідкових даних щодо значень ентропії відповідних речовин обчислити ентропію реакції конверсії метану.
Розв’язок. Реакція конверсії метану проходить згідно з рівнянням
СН4(г) + Н2О(г) = СО(г) + 3Н2(г).
Виходячи з співставлення коефіцієнтів, можна якісно оцінити змінення ентропії: вона повинна зростати[TEX](\Delta{S}^0_{298}>0)[/TEX], оскільки в результаті реакції утворюється більша кількість газових молекул, ніж вступає в реакцію. Це свідчить про посилення невпорядкованості системи і, як наслідок, – про збільшення ентропії. Ентропія – функція стану, тому її змінення визначається тільки початковим і кінцевим станами системи:
[TEX]\Delta{S}^0_{298}=S^0_{298}(СО)+3S^0_{298}(Н_2)-S^0_{298}(СН_4)-S^0_{298}(Н_2О)=197,54+3\cdot{130,58}-186,19-188,70= +214,39[/TEX]Дж/К.
Як видно із прикладу, ентропія системи в результаті реакції зросла,[TEX]\Delta{S}>0[/TEX]

