

- 7.1 Поняття про енергію активації хімічної реакції
- 7.2 Oсновні поняття та визначення хімічної кінетики
- 7.3 Кінетична класифікація гомогенних хімічних реакцій
- 7.4 Способи визначення порядку реакції
- 7.5 Поняття про фотохімічні та радіаційно-хімічні реакції
- 7.6 Вплив температури на швидкість хімічної реакції
Ключові терміни:
Енергія активації, Реакції другого порядку, Реакції нульового порядку, Реакції першого порядку, Реакції третього порядку, Рівняння Арреніуса, Фотохімічні реакції, Час напівперетворення, Швидкість хімічної реакції, правило Вант-ГоффаНа попередніх лекціях йшлося про основні закономірності хімічних процесів з точки зору хімічної термодинаміки. Такий підхід дозволяє на основі вивчення енергетичних властивостей системи та її ентропії визначити рівноважний стан, тобто можливий кінцевий результат хімічної взаємодії. При цьому дуже часто отриманий із термодинамічного розгляду висновок щодо перебігу процесу не означає, що дана хімічна реакція в дійсності відбувається за умов, що розглядаються. Наприклад, нас цікавить перетворення однієї кристалічної модифікації вуглеводню – алмазу – в іншу його модифікацію – графіт:
- C (алмаз)
 С (графіт).
С (графіт).
Термодинамічне вивчення такої системи показує, що при стандартних умовах графіт більш стійкий, ніж алмаз: Gгр = -1,71 кДж/моль, Gал = 1,18 кДж/моль. Тому в зазначених умовах алмаз повинен самодовільно перетворюватися у графіт, оскільки Δ G = Gгр – Gал = = -2,89 кДж/моль. У дійсності цього перетворення не відбувається. Така уявна суперечність теоретичних розрахунків і практичних спостережень пояснюється обмеженістю термодинамічного методу, оскільки він розглядає лише початковий та кінцевий стан системи.
Дійсно, за звичайних умов вільна енергія графіту менша за вільну енергію алмазу. Справа в тому, що перетворення кристалічної ґратки алмазу в ґратку графіту обов´язково відбувається через якісь проміжні структури, що відрізняються від обох ґраток (рис.7.1).
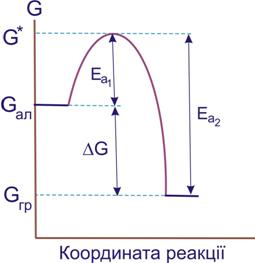
Рисунок 7.1. До визначення енергії активації
Утворення проміжних структур пов´язане з витратами енергії, що досягає G*. Тому фактичне змінювання енергії у процесі реакції буде зображено кривою з максимумом. Висоту максимуму в порівнянні з енергією вихідної речовини називають енергією активації даної хімічної реакції Еа. Іншими словами, термодинамічно можливий перехід системи з початкового стану в кінцевий пов´язаний з подоланням енергетичного бар’єра, що уповільнює реальний перебіг процесу, особливо за низьких температур.
Можна навести багато прикладів, коли термодинамічно можливі процеси практично не здійснюються через дуже малі швидкості. Так, реакція взаємодії водню та кисню
- 2Н2 + О2 2Н2О
супроводжується дуже великою втратою вільної енергії ΔG298= - 455,6 кДж/моль, тобто процес повинен відбуватися самодовільно. Однак відомо, що суміш цих газів (гримуча суміш) може перебувати значний час без помітного утворення води. Причиною цього є високий енергетичний бар'єр, висока енергія активації процесу.
Зробимо визначення цієї величини. Енергія активації – це та надлишкова кількість енергії (у порівнянні з середньою величиною), яку повинні мати молекули в момент зіткнення, щоб прореагувати. Ця надлишкова енергія може бути в молекулах у різних формах. Зокрема: 1) підвищена кінетична енергія поступового або обертального руху молекули; 2) підвищена енергія внутрішньо молекулярних коливань атомів; 3) підвищена енергія руху електронів тощо.
Як бачимо з рис. 7.1, енергії активації прямої (Еа1) і зворотної (Еа2) реакцій різні. Наприклад, для реакції
- Н2 + І2 = 2НІ
- Еа1 = 166,0 кДж/моль, а Еа2 = 185,6 кДж/моль.
З вищезазначеного зрозуміло, що вивчення хімічних систем з однієї лише термодинамічної точки зору є недостатнім. Не менш важливе вивчення в іншому аспекті – з точки зору швидкостей процесів та їх механізмів, тобто з точки зору кінетики.
Швидкість хімічної реакції W – це змінювання кількості будь-якої речовини (або вихідної, або продукту) ni за одиницю часу t й за одиниці реакційного простору R:
-
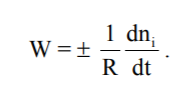
. (7.1)
Цей вираз використовують зі знаком “плюс”, коли швидкість хімічної реакції визначають за змінюванням кількості продукту реакції (яка є позитивною), або зі знаком “мінус”, коли швидкість реакції визначають за змінюванням кількості будь-якої вихідної речовини (яка є негативною). В обох випадках швидкість реакції залишається позитивною.
Наведене визначення швидкості хімічної реакції є загальним, воно справедливе для будь-яких реакцій. Якщо реакція гомогенна та проходить в об’ємі, то реакційним простором є об’єм (R ≡ V). Якщо реакція гетерогенна і проходить на поверхні розподілу фаз, то реакційним простором є ця поверхня (R ≡ S).
Найбільш теоретично описаними з точки зору хімічної кінетики є гомогенні реакції в ізольованих або закритих системах. Тому розглянемо на прикладі таких систем основні закономірності хімічної кінетики. Це можуть бути або газові реакції, або реакції між розчинами (за умови, що реагуюча суміш є однією фазою). У цьому випадку для швидкості хімічної реакції можна записати
-
.
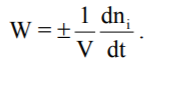 (7.2)
(7.2)
Якщо V = const, то враховуючи, що ni/V = Ci, де Сі – концентрація будь-якого компонента, можна записати
- . (7.3)
Вирішимо ще одне принципове питання. Нехай у системі проходить гомогенна реакція за участю речовин А, В, D, F:
- аА + вВ
 dD + fF,
dD + fF,
а концентрації речовин відповідно дорівнюють СА, СВ, СD i CF. Тоді для однозначного визначення швидкості реакції достатньо стежити за змінюванням концентрації однієї будь-якої речовини. Змінювання концентрацій усіх інших учасників реакції можна знайти із співвідношення
-
. (7.4)
![]() Уведемо поняття про елементарну реакцію. Такою називається реакція, що проходить у відповідності до її рівняння, тобто за рахунок одночасного зіткнення молекул, що записані в лівій частині рівняння. Слід сказати, що з цих позицій у елементарній реакції можуть брати участь одна, дві або (спостерігається дуже рідко) три молекули. Тому елементарні реакції підрозділяються на моно-, бі- та тримолекулярні.
Уведемо поняття про елементарну реакцію. Такою називається реакція, що проходить у відповідності до її рівняння, тобто за рахунок одночасного зіткнення молекул, що записані в лівій частині рівняння. Слід сказати, що з цих позицій у елементарній реакції можуть брати участь одна, дві або (спостерігається дуже рідко) три молекули. Тому елементарні реакції підрозділяються на моно-, бі- та тримолекулярні.
Математичне рівняння, що пов’язує швидкість реакції з концентраціями речовин, які реагують, називають кінетичним рівнянням даної хімічної реакції. Слід зазначити, що за зовнішнім виглядом рівняння хімічної реакції не можна зробити висновок про її кінетичне рівняння (нагадаємо, що в хімічній термодинаміці за зовнішнім виглядом рівняння можна провести розрахунок різних величин, наприклад, константи рівноваги). Проілюструємо вищезазначене. Реакції водню з парами йоду або брому виражаються однаковими стехіометричними рівняннями. Однак кінетичні рівняння для них різні, саме:
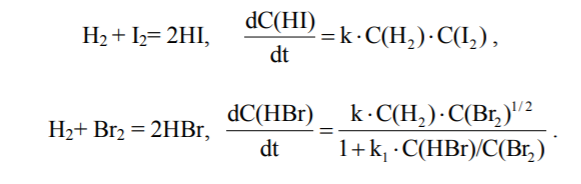
Це пояснюється тим, що перша реакція проходить як елементарна, у той самий час утворення HBr є результатом складної ланцюгової реакції, механізм якої можна подати так:
- Br2
 2Br,
2Br,
- Br + H2 HBr + H,
- H + Br2 HBr + Br.
Таким чином, кінетичне рівняння хімічної реакції може бути встановлене виключно експериментальним шляхом.
Основним законом хімічної кінетики є постулат, що випливає з великої кількості експериментальних даних і виражає залежність швидкості від концентрації. Цей закон, що отримав назву закону діючих мас, формулюється так:
Швидкість хімічної реакції в кожний момент часу пропорційна добутку концентрацій реагуючих речовин, піднесених до деяких ступенів.
Якщо емпіричне рівняння реакції
- аА + bB + dD продукти,
то математичну формулу основного закону можна подати у вигляді
-
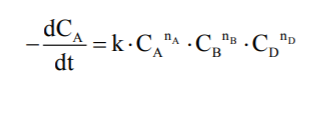
. (7.5)
У цьому рівнянні коефіцієнт пропорційності k не залежить від концентрацій. Він дуже різний для різних реакцій і значно залежить від температури. Називають k константою швидкості реакції. Фізичний вміст k легко знайти, ураховуючи, що СА = СВ = СD = 1 моль/л. Тоді W = k, тобто k чисельно дорівнює швидкості реакції за концентраціями реагуючих речовин, що дорівнюють одиниці. Показники ступенів nA, nB, nD, як правило, є позитивними цілими числами, але іноді бувають дробові і навіть негативні показники. Поява цих ускладнень пояснюється більш або менш складним механізмом реакції.
З точки зору хімічної кінетики усі елементарні реакції можна класифікувати на реакції нульового, першого, другого та третього порядків. На відміну від молекулярності порядок реакції – величина формальна й дорівнює сумі показників степенів у кінетичному рівнянні. Із співвідношення (7.5) загальний порядок n даної реакції дорівнює
- n = nA + nB + nD.
При цьому величини nA, nB, nD називаються приватними порядками реакції за реагентами А, В і D відповідно. Отже, загальний порядок реакції n дорівнює сумі порядкув за кожним з реагентів.
Для елементарних реакцій nA = a , nB = b, nD = d.
Можливі такі типи кінетичних рівнянь елементарних реакцій:
-
n = 1 A продукти W = kCA;
-
n = 2 2A продукти W = kCA2;
- A1 + A2 продукти W = k·C(A1)·C(A2);
-
n = 3 3A продукти W = kCA3;
- A1 + A2 продукти W = k·C2(A1)·C(A2);
- A1 + A2 + A3 продукти W= k·C(A1)·C(A2)·C(A3);
-
n = 0 A продукти W = k.
Реакції нульового порядку – це реакції, які відбуваються при надлишку реагентів так, що змінювання концентрації не впливає на швидкість реакції.
Знайдемо основні кінетичні характеристики й рівняння для гомогенних хімічних реакцій різного порядку в закритих системах при постійності об´єму та температури
Реакції нульового порядку. Трапляються рідко. Прикладом таких є реакція розкладання N2O5 у газовій фазі за наявності твердого N2O5, коли постійна концентрація реагенту в газовій фазі підтримується сталою за рахунок випарювання твердого оксиду. Для реакцій нульового порядку
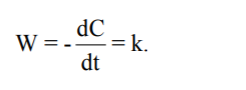
Розв´яжемо це рівняння, розділивши змінні
Після інтегрування отримаємо
С = - k•t + B,
де В – константа інтегрування. Знайдемо її, ураховуючи ту початкову умову, що в момент t = 0 концентрація дорівнює початковій С = С0, звідси С0 = В і
C = С0 – k•t.
Отже, у реакціях нульового порядку концентрація лінійно зменшується з часом (рис. 7.2,а).
Константу швидкості реакції нульового порядку можна обчислити за рівнянням
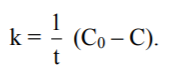
.
Її розмірність (моль/л)·с-1).
Поряд з константою швидкості для характеристики реакцій часто використовують величину, що називається часом напівперетворення t1/2 .
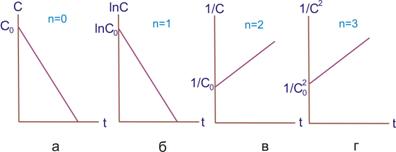
Рисунок 7.2. Змінювання концентрації з часом у реакціях нульового (а), першого (б), другого (в) і третього (г) порядків
Час напівперетворення – проміжок часу, за який реагує половина вихідної кількості речовини, тобто при t =1/2t C = C0/2, звідси
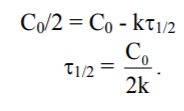
.
Отже, для реакції нульового порядку час напівперетворення пропорційний початковій концентрації вихідної речовини.
Реакції першого порядку. До елементарних реакцій першого порядку належать такі, які можна подати у вигляді
- A
 продукти.
продукти.
Частіше всього це реакції розкладу. Наприклад, реакція розкладу ацетону
CH3OCH3 ⇒ CH4 + H2 + CO.
Кінетичне рівняння для реакцій першого порядку має вигляд
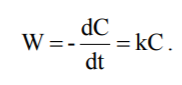
.
Проінтегруємо його, розділивши змінні:
- ln C = - kt + B.
Для визначення постійної інтегрування припустимо, що в початковий момент реакції t = 0 концентрація вихідної речовини була С0, тоді
- ln C0 = B,
- отже,
- ln C = - kt + lnC0
Константа швидкості такої реакції має розмірність (с)-1. Величина, зворотна константі швидкості реакції першого порядку, має розмірність часу й називається середньою тривалістю життя окремої частинки.
Для реакції першого порядку характерна лінійна залежність lnC від t (рис. 7.2,б).
Знайдемо час напівперетворення для реакції першого порядку. Для t = 1/2t; C = C0 /2.
Тому
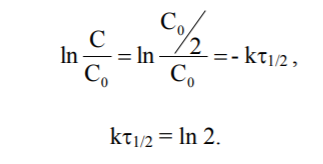
-
.
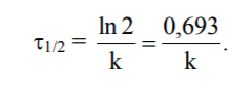
Бачимо, що час напівперетворення визначається виключно значенням константи швидкості. Так, для наведеної реакції розкладання ацетону k = 4,27 × 10-4 c-1. Отже,
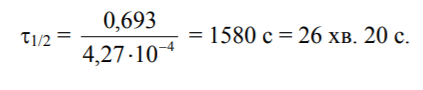
До істинно молекулярних або, краще сказати, моноатомних процесів першого порядку відносять усі численні перетворення радіоактивних речовин. У табл. 7.1 наведені деякі дані для таких реакцій.
Таблиця 7.1. Кінетичні характеристики радіоактивного розпаду деяких ізотопів
|
Ізотоп |
k, c-1 |
|
|---|---|---|
|
222Ra 226Ra 238U 214Po |
2,1×10-6 1,35×10-11 4,88×10-18 4,62×103 |
3,8 доби 1620 років 4,5×109 років 1,5×10-4 с |
Класичним прикладом реакції першого порядку є реакція інверсії тростинного цукру (сахарози)
| С12Н22О11 + Н2О |
C6H12O6 + | C6H12O6 |
| сахароза | глюкоза | фруктоза |
Кінетичне рівняння цієї реакції має вигляд
-
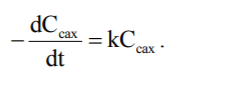
.
У реакції беруть участь ще вода й кислота (іони Н+), однак концентрація кислоти є постійною (іони Н+ – каталізатор), а концентрація води, що міститься в дуже великому надлишку, теж практично постійна. Реакція інверсії зручна для вивчення тим, що в ході її змінюється кут обертання площини поляризації. Сахароза обертає площину поляризації управо, а суміш глюкози та фруктози – уліво. Поміщаючи розчин сахарози у трубку поляриметра, можна стежити за проходженням реакції, не перериваючи її. При кінетичних розрахунках використовується пропорційний зв´язок між кутом обертання та концентрацією обертальної речовини.
Реакції другого порядку. Такі проходять за участю двох частинок (молекул, атомів). Якщо в елементарній реакції беруть участь дві однакові частинки. Для реакції другого порядку спостерігається лінійна залежність 1/с від t (рис. 7.2, в).
2А ![]() продукти,
продукти,
то кінетичне рівняння має вигляд
-
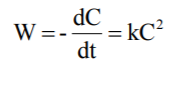
,
- де С – концентрація речовини А. Інтегрування цього рівняння приводить до
-
.
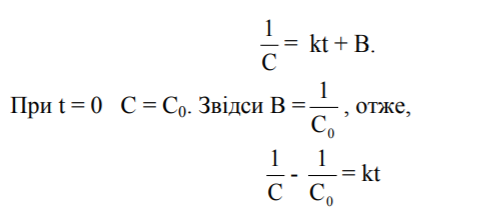
- або
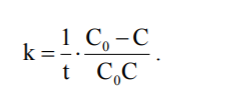
-
.
- Розмірність константи швидкості реакції другого порядку (моль/л)·с-1, тобто на відміну від реакцій першого порядку в розмірності є не тільки час, але й концентрація. Тому для реакцій другого порядку (та вище) не можна при розрахунках замінювати концентрацію на пропорційні їй величини.
-
Для реакції другого порядку
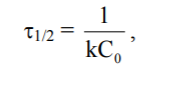
,
-
тобто чим більша початкова концентрація, тим менший час напівперетворення.
Якщо в елементарній реакції другого порядку реагують дві різні частинки
А1 + А2 продукти, - причому концентрації речовин А1 та А2 різні, то

- Другий порядок мають, наприклад, такі реакції::
-
2HI ⇒ H2 + I2,
2NO2 ⇒ 2NO + O2.
Окислення бромоводню киснем за схемою
4HBr + O2 → 2Br2 + 2H2O
теж є реакцією другого порядку, незважаючи на те, що відповідно до рівняння реакції з однією молекулою кисню взаємодіють чотири молекули HBr. Розбіжність між коефіцієнтами в рівнянні реакції і математичним виразом для її швидкості
ϑ=k•C(O2)•C(HBr)
пов’язана з тим, що у дійсності механізм реакції складається з трьох стадій:
1) НВr + О2 ⇒ HО2Вr (лімітуюча, найповільніша стадія),
2) НО2Вr + НBr ⇒ 2НОВr,
3) 2НОBr + 2НВr ⇒ 2Н2О + 2Вr2,
____________________________________________
4НВr + О2 ⇒ 2Н2О + 2Вr2.
Перша стадія у розглянутому механізмі – найповільніша, а друга і третя – дуже швидкі, тому вони практично не впливають на тривалість реакції. Швидкість реакції у цілому визначається найповільнішою стадією, яка називається лімітуюча стадія. Саме тому взаємодія між НВr і О2, швидкість якої однаково залежить від концентрацій обох реагентів, належить до реакцій другого порядку.
-
Реакції третього порядку. Їх можна подати у вигляді:
- 3А продукти,
- 2A1+ A2 продукти,
- A1 + A2 + A3 продукти.
-
Оскільки реакції третього порядку спостерігаються дуже рідко й не становлять значного практичного інтересу, розглянемо їх кінетику тільки для випадку рівності концентрацій усіх реагуючих речовин.
Тоді
-
.
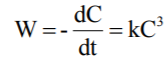
-
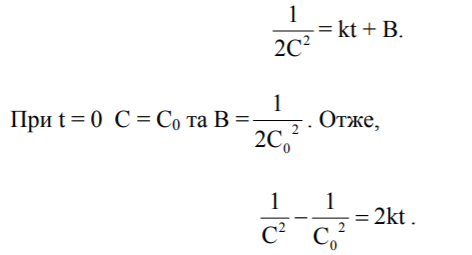
.
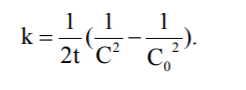
-
.
- Константа швидкості реакції третього порядку має розмірність (моль/л)-2·с-1.
- Період напіврозпаду для реакцій третього порядку:
-
-
-
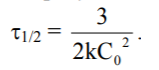
- Для моделювання хіміко-технологічних процесів корисно ввести поняття формально простих реакцій . До них відносять будь-які реакції, для яких кінетичне рівняння може бути подане наближено у вигляді степеневої залежності
-
-
- W = kC1n(1) · C2n(2) · C3n(3) …
-
Наприклад, такою є складна реакція:
-
3СН3ОН + 2Н2CrO4 + 6HCl
3CH2O + 2CrCl3 + 8H2O. - Для цієї реакції
- W = kC(CH3OH) · C(H2CrO4) · C2(HCl).
-
Як бачимо, степені для формально простої реакції не збігаються зі стехіометричними коефіцієнтами. У загальному випадку для таких реакцій показники степенів можуть бути дробовими і набувати значень, більших від трьох.
-
-
-
- 3А
7.4 Способи визначення порядку реакції
Порядок реакції, у якому бере участь одна речовина, можна визначити, користуючись даними експериментів для двох різних початкових концентрацій С01 і С02. Тоді
-
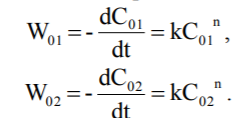
,
-
.
Логарифмуємо ці рівняння й отримуємо
-
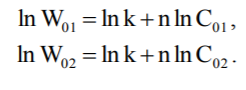
,
Віднімаємо рівняння одне із одного й отримуємо
-
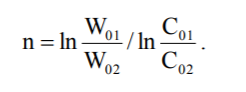
.
Для реакцій, у яких беруть участь дві вихідні речовини
- аА + вВ продукти,
- кінетичне рівняння має вигляд
- W = k CAn(A) · CBn(B) ,
і задача зводиться до визначення порядків реакції за кожним з реагентів nA i nB, оскільки n = n(A) + n(B).
Для цього проводять дві серії експериментів: перша – при постійній (або надлишковій) концентрації речовини А, друга – при постійній (або надлишковій) концентрації речовини В. У обох модифікаціях концентрація однієї з речовин не буде впливати на швидкість реакції. Маємо
де kB = kCAn(A) i kA = kCBn(B). Після логарифмування отримуємо
- lnW1 = lnkB + n(B) lnCB,
- lnW2 = lnkA + n(A)lnCA.
Дослідні точки наносимо на графік з координатами lnW – lnC. Отримуємо дві прямі (рис.9.3). n(B)=tgφ1, n(A)=tgφ2, n=tgφ1+tgφ2. Величини відрізків, що відсікаються прямими на осі ординат, дозволяють визначити k.
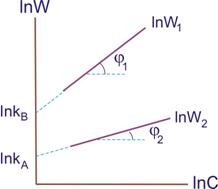
Рисунок 7.3. До визначення порядку реакції
Подані способи називаються диференціальними. Крім того, існують інтегральні способи визначення порядку реакції за експериментальними даними. Вони базуються на перевірці виконання кінетичних залежностей для реакцій різних порядків. Розглянемо ці способи.
- Графічний спосіб. Як бачимо з рис. 7.2 та проведених нами розв´язань кінетичних рівнянь для реакцій різних порядків, лінійна залежність f(c) – t реалізується у різних координатах. Для визначення порядку реакції будують усі чотири залежності (C-t; lnC-t; 1/C-t; 1/C2-t). Якщо, наприклад, графік, побудований за дослідними даними, виявився прямолінійним у координатах 1/С – t , то порядок реакції за розглянутою речовиною дорівнює двом.
- Спосіб підстановки. Для різних моментів часу за формулами для реакцій можливих порядків розраховують значення константи k. Якщо розрахункові значення k за однією з формул не залежать від часу, то це означає, що ця формула відповідає реакції, що вивчається експериментально.
-
Спосіб часу напівперетворення. Послідовно обчислюємо
для декількох концентрацій за формулами для різних порядків, щоб встановити залежність
Фотохімічні реакції це реакції, які відбуваються при дії світла на речовину. В основу фотохімії покладено два закони. Згідно з першим законом (закон Гротгуса-Дрейпера)
фотохімічні реакції можуть бути викликані тільки тією частиною падаючого світла, яка поглинається реагуючими речовинами, тобто є доповненням до їх кольору.
Другий закон фотохімії (Ейнштейна) стверджує:
кожен квант світла, що поглинувся, викликає перетворення однієї молекули.
Механізм фотохімічних реакцій складається з декількох стадій. Початкова стадія:
- збудження молекул М + hν
 M*,
M*,
- дисоціація молекул М2 + hν 2М• ,
- іонізація молекул М + hν М+ + е.
Активована частинка існує ~ 10-8 с, після чого втрачає надлишок енергії. Це звичайні фотохімічні процеси. Наприклад:
- флуоресценція М* A + hν ,
- дисоціація М* A1 + A2,
- внутрішня перебудова М* L,
- реакція М* + В C.
Тоді, коли взаємодія активованих частинок, що утворюються в первинних процесах, з іншими частинками системи приводить до хімічних перетворень, починається остання стадія - стадія вторинних процесів. Для їх проходження не треба освітлення, тому їх ще називають темновими процесами.
Для кількісної характеристики фотохімічних реакцій введене поняття квантового виходу γ:
- γ = кількість молекул, що прореагували /кількість поглинутих квантів.
Швидкість фотохімічної реакції пропорційна потужності світлового потоку Фе, довжині світлової хвилі λ та квантовому виходу γ:
-
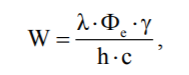
,
- де h - стала Планка; с - швидкість світла у вакуумі.
Залежно від величини γ розрізняють такі випадки:
- γ = 1. Реакції є чисто фотохімічними. Їх дуже мало і звичайно вони використовуються в лабораторній практиці для визначення кількості поглинутих квантів світла.
- γ < 1. У цьому разі частина поглинутої енергії розсіюється при зіткненні активованої частинки з молекулами розчинника (такі реакції проходять у розчинах).
- γ > 1. Це ланцюгові фотохімічні реакції. Їх квантовий вихід може набувати дуже великих значень. Наприклад, для реакції
Н2 + Cl2→ 2HCl, γ ≈ 105.
Серед численних фотохімічних реакцій особливе значення має реакція асиміляції вуглецю рослинами, без якої не може існувати життя на Землі:
- 6СО2 + 6Н2О = С6Н12О6 + 6О2, γ < 1, ΔG = 2860 Кдж/моль.
-
Незважаючи на позитивне значення енергії Гіббса, ця реакція відбувається у листках рослин завдяки енергії, що отримується ззовні від сонця.
Фотографія є найважливішим практичним застосуванням фотохімічних процесів. Основою фотографічного процесу є здатність галогенідів срібла розкладатися під дією світла з виділенням металічного срібла
- AgBr + hν ⇒Ag + Br.
-
Радіаційно-хімічні реакції (радіоліз) проходять під дією на речовину випромінювань високих енергій. До іонізуючих випромінювань належать рентгенівські та гамма-випромінювання, а також пучки електронів, протонів, нейтронів, α -частинок тощо.
Радіоліз значно відрізняється від фотолізу. Поглинання випромінювань, що мають значно більшу енергію, ніж видимі інфрачервоні та ультрафіолетові промені, спричиняє збудження або відрива електронів від зовнішніх оболонок атомів. Відбувається іонізація речовини, що руйнує зв’язки між атомами в молекулах і приводить до появи хімічно активних частинок - радикалів, іонів, збуджених атомів та молекул. Внаслідок цього можуть утворюватися численні хімічні сполуки.
Утворення вільних радикалів та атомів із ненасиченою валентністю, що проходить при опромінюванні, було використане для процесів полімеризації стиролу, акрилонітрилу та інших речовин. Під дією γ-випромінювання в поліетилені та інших полімерах утворюються додаткові поперечні зв’язки, що збільшує міцність і стійкість полімеру.
Як відомо, швидкість більшості хімічних реакцій, за незначним винятком (наприклад, 2NO + O2![]() 2NO2), стрімко збільшується з підвищенням температури. Тому нагрівання є прийомом, який дуже поширений у хімічній практиці. Кількісно оцінити вплив температури на швидкість (а фактично на константу швидкості) реакції можна кількома шляхами. Для реакцій у розчинах, що відбуваються за порівняно невеликих температур, можна використовувати напівкількісне емпіричне правило Вант-Гоффа:
2NO2), стрімко збільшується з підвищенням температури. Тому нагрівання є прийомом, який дуже поширений у хімічній практиці. Кількісно оцінити вплив температури на швидкість (а фактично на константу швидкості) реакції можна кількома шляхами. Для реакцій у розчинах, що відбуваються за порівняно невеликих температур, можна використовувати напівкількісне емпіричне правило Вант-Гоффа:
при підвищенні температури на 10° швидкість реакції збільшується у 2-4 рази.
Використовуючи поняття температурного коефіцієнта швидкості реакції γ, можна розрахувати зміну швидкості зa будь-якої зміни температури:
- [TEX]\frac{w_{2} }{w_{1} } =\gamma ^{t_{2}-t_{1}/10 } [/TEX]
,
- де t2-t1 - кількість десятків градусів, на яку змінилася температура.
Більш точно позначає залежність константи швидкості реакції від температури рівняння, яке запропоноване в 1889 р. шведським вченим С.Арреніусом (рівняння Арреніуса):
- k = k0exp(- Ea/RT),
- де k0 - передекспоненціальний множник, а Еa - енергія активації реакції.
Згідно з теорією Арреніуса (теорії активних зіткнень) для проходження реакції необхідно:
- щоб відбулися зіткнення частинок системи;
-
щоб зіткнення частинок дійсно привели до хімічного перетворення, оскільки ефективні не всі зіткнення, а тільки ті, у яких беруть участь “активні” молекули, що мають енергію, більшу за Еа.
- 2HI
 I2 + H2
I2 + H2
- 2HI
- взаємодіючі молекули при зіткненні повинні бути певним чином зорієнтовані у просторі, тобто повинні утворювати конфігурацію, яка найбільш придатна для розриву одних зв’язків та виникнення інших.
Проілюструємо важливість цього положення простим розрахунком. Константа швидкості реакції [TEX]2HI\leftrightarrow{I_2+H_2}[/TEX] при [TEX]100^oC[/TEX] дорівнює [TEX]k=8,83\cdot{10^{-16}}(моль/л)^{-1}\cdot{c^{-1}}[/TEX]. Щоб зробити наочним це значення розрахуємо час напівперетворення для цієї реакції другого порядку при С0 = 1 моль/л:
- [TEX]\tau_{1/2}=\frac{1}{kC_0}=\frac{10^{16}}{8,83\cdot{3600}\cdot{24}\cdot{365}}=3,6\cdot{10^7}[/TEX]лет.
Таким чином, при 1000С половина вихідного йодистого водню розклалася б за 36 мільйонів років. Причина цього в тому, що за даної температури дуже мала частка активних молекул. Табл. 7.2 ілюструє залежність частки активних молекул від температури для різних значень енергії активації.
Чим більші значення Еа, тим сильніше швидкість реакції залежить від температури. Однак якщо частка активних молекул більша за 10-7, то така реакція проходить дуже швидко (вибух), а якщо менша за 10-18 , то реакція проходить дуже повільно.
Таблиця 7.2. Частка активних молекул
|
Т, К |
Енергія активації, Еа, Кдж/моль |
|||
|---|---|---|---|---|
|
5 |
50 |
10 |
150 |
|
|
300 600 1000 1500 |
0,13 0,37 0,55 0,67 |
2×10-9 4×10-5 2×10-3 2×10-2 |
4×10-18 2×10-9 6×10-6 3×10-4 |
8×10-27 9×10-14 1×10-8 6×10-6 |
Рівняння Арреніуса набирає вигляду
-
k = A· P e – Еакт/RT,
-
де Р - стеричний множник, який визначає просторову орієнтацію взаємодіючих молекул (Р < 1); А - стала (А×Р= =k
-
Було наведено, що Р = ехр (Sa/R), де Sa - ентропія активації. Тому
k = Z · e-Ea/RT · eSa/R,
де Z - стала. Отже, можна казати про ентальпійний (exp[-Ea/RT]) та ентропійний (exp Sa/R) чинники. Проте ентропійний чинник не залежить від температури. Його роль особливо помітна для реакцій, у яких беруть участь складні молекули, взаємна орієнтація яких повинна бути такою, щоб одна до одної підходили певні ділянки взаємодіючих молекул. Наприклад, для реакції
- Н2 + І2⇒2HI
- P ~ 1, а для реакції
- С2Н5І + (С2Н5)3N ⇒ (C2H5)4NI
- P ~ 10-6.
-
Для знаходження констант рівняння Арреніуса дані визначення швидкості реакції при різних температурах подають у логарифмічному вигляді:
-
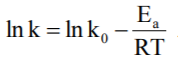
Потім у координатах ln k - 1/T будують графік прямої (рис. 7.4), тангенс кута нахилу якої дорівнює -Еа/R, а відрізок, що відсікається продовженням прямої на осі ординат, дорівнює ln k0 (зручніше розрахувати рівняння цієї прямої на ЕОМ і отримати її коефіцієнти).

Рисунок 7.4. Графічне визначення енергії активації
Слід зазначити, що хоча швидкість більшості реакцій підвищується із збільшенням температури, це підвищення не завжди монотонне й іноді на кривих залежності швидкості від температури спостерігаються максимуми або мінімуми. Це пов’язано із складним механізмом таких реакцій, наявністю каталізаторів та низкою інших причин.

