

- 4.1 Загальні поняття хімічної кінетики
- 4.2 Швидкість хімічної реакції
- 4.3 Каталіз
- 4.4 Швидкість гетерогенних реакцій
- 4.5 Необоротні та оборотні хімічні реакції. Умова хімічної рівноваги
- 4.6 Константа хімічної рівноваги
- 4.7 Вплив зовнішніх чинників на хімічну рівновагу
- 4.8 Приклади розв'язання типових задач
Ключові терміни:
активні молекули, активований комплекс, гетерогенні реакції, гомогенні реакції, дифузійний контроль, дифузійний шар, елементарні стадії, енергія активації, ефективні зіткнення, загальний порядок реакції, закон Гульдберга-Вааге, закон діючих мас, закон діючих мас для стану рівноваги, змішаний контроль, зміщення хімічної рівноваги, зсув хімічної рівноваги, каталіз, каталізатор, каталітичні отрути, константа рівноваги, константа швидкості, кінетичне рівняння, кінетичний контроль, лімітуюча стадія, лімітуюча стадія, механізм реакції, молекулярність реакції, наслідки принципу Ле-Шательє, необоротні реакції, оборотні реакції, порядок реакції за реагентом, правило Вант-Гоффа, принцип Ле-Шательє, промотор, рівноважні концентрації, рівняння Арреніуса, селективність каталізаторів, середня швидкість реакції, стеричний фактор, сучасне формулювання закону діючих мас, температурний коефіцієнт, хімічна кінетика, хімічна рівновага, швидкість гетерогенної реакції, швидкість реакції, інгібітор, істинна швидкість реакції
4.1 Загальні поняття хімічної кінетики
Хімічна термодинаміка дозволяє передбачити принципову можливість чи неможливість самочинного перебігу реакції, а також розрахувати рівноважні концентрації реагуючих речовин. Однак цього недостатньо для визначення швидкості і механізму реакції та керування процесом. Тривалість реакції найчастіше не пов’язана зі значенням енергії Гіббса. Наприклад, термодинамічні розрахунки свідчать, що можливість самочинного утворення води з простих речовин за схемою
1) Н2 + ½ О2 → Н2О(р), (ΔG0298 = −237,2 кДж/моль),
значно вища порівняно з утворенням води за реакцією нейтралізації
2) Н+ + ОН‾ → Н2О(р) (ΔG0298 = −79,9 кДж/моль).
Незважаючи на те, що для реакції (1) енергія Гіббса має менше значення, за звичайних умов вона без наявності каталізатора не відбувається. І навпаки, реакція (1) перебігає практично миттєво. Такі якісні та кількісні особливості процесів, що проходять протягом деякого часу, пояснює хімічна кінетика.
Хімічна кінетика – це розділ хімії, який вивчає швидкість і механізми перебігу хімічних реакцій та їх залежність від окремих чинників. Хімічна кінетика вирішує дві конкретні задачі, першою з яких є визначення механізму реакції.
Механізм реакції – це сукупність і послідовність елементарних стадій, через які проходить хімічна реакція від вихідних речовин до кінцевих продуктів.
Звичайне рівняння реакції містить інформацію тільки про склад і кількість речовин, що вступають у реакцію та утворюються внаслідок неї, але не відображає реальних процесів, які відбуваються в дійсності, тобто не описує елементарні стадії.
Елементарні стадії – це проміжні одиничні процеси протягом хімічної реакції, які не можуть бути розділені на простіші акти хімічної взаємодії і які включають зіткнення реагуючих частинок, розрив зв'язків у вихідних сполуках, утворення проміжних сполук і взаємодію між ними, виникнення нових зв'язків і одержання продуктів реакції.
Встановлення механізму пов'язано з класифікацією реакцій за молекулярністю.
Молекулярність реакції – це характеристика, яка визначається кількістю молекул, що беруть участь в елементарній стадії при взаємодії чи перетворенні частинок.
За молекулярністю розрізнюють такі реакції:
- мономолекулярні (рис. 4.1 а), при яких відбувається перетворення однієї молекули (ізомерізація, дисоціація тощо), наприклад: I2 → 2I;
- бімолекулярні реакції (рис. 4.1 б), при яких здійснюється перетвореня двох частинок (молекул, іонів, радикалів, атомів). Наприклад, взаємодія між атомом водню, який має неспарений електрон (на схемі позначений точкою) з молекулою хлору:
Н• + Cl2→ HCl + Cl•;
- тримолекулярні реакції, які проходять при одночасному зіткненні трьох частинок (рис. 4.1 в), наприклад:
2NO + H2→ N2O + H2.
Доведено, що одночасне зіткнення більше трьох молекул практично неможливе. Наявність у рівнянні реакції великих стехіометричних коефіцієнтів (коли їх сума перебільшує 3) однозначно вказує на складний механізм, який включає декілька елементарних актів.
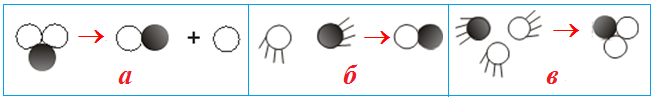
Рисунок 4.1 – Моделі реакцій: а) мономолекулярні реакції; б) бімолекулярні реакції; в) тримолекулярні реакції
Другою задачею хімічної кінетика вважається кількісний опис хімічної реакції за допомогою кінетичного рівняння.
Кінетичне рівняння – це математичний вираз, який описує залежність швидкості реакції від концентрації вихідних речовин і дає можливість визначати змінення кількостей реагентів і продуктів реакції протягом її перебігу.
4.2 Швидкість хімічної реакції
Швидкість хімічної реакції характеризує інтенсивність хімічного процесу, тобто кількість елементарних актів перетворення частинок протягом певного часу. Але взаємодія між частинками може відбуватися тільки при їх безпосередньому контакті, який певним чином визначається особливостями реакційного середовища, або реакційного простору.
За ознакою фазового складу реакційного простору в хімічній кінетиці розрізнюють:
-
гомогенні реакції, які перебігають в одній фазі одночасно по всьому реакційному простору, причому, між окремими речовинами в реакційній системі відсутня межа поділу (рис. 4.2 а), наприклад, реакція в рідкому середовищі:
2K2CrO4(р) + H2SO4(р) → K2Cr2O7(р) + K2SO4(р) + H2O(р);
-
гетерогенні реакції, в яких речовини в реакційній системі відокремлюються одна від одної поверхнею поділу фаз, наприклад (рис.4.2 б):
2Na(тв) + 2H2O(р) → 2NaOH(р) +H2↑(г).

Рисунок 4.2 – Ілюстрація до класифікації хімічних реакцій за фазовим складом:
а) гомогенна реакція по перетворенню K2CrO4 → K2Cr2O7 (у пробірці ліворуч – вихідний розчин K2CrO4 жовтого кольору, праворуч – продукт реакції K2Cr2O7 оранжевого кольору); б) гетерогенна реакція між Na і Н2О (додавання індикатору фенолфталеїну зумовлює появу малинового забарвлення, що вказує на утворення лугу NaOH)
Швидкість реакції ϑ – це фізична величина, яка визначається кількістю речовини, що вступає в реакцію чи утворюється внаслідок реакції за одиницю часу в одиниці реакційного простору, тобто в одиниці об’єму для гомогенних реакцій чи на одиниці площі реакційної поверхні – для гетерогенних.
Однак відношення кількості речовини [TEX]\nu[/TEX] до одиниці об’єму [TEX]V[/TEX] – це молярна концентрація [TEX]C[/TEX], яка визначається з рівняння [TEX]C=\nu/V[/TEX]. Тому швидкість гомогенної реакції дорівнює зміненню концентрації вихідної сполуки чи продукту реакції протягом часу. Завдяки стехіометричному співвідношенню речовин у хімічній реакції, контроль за зміненням концентрації звичайно здійснюється тільки для однієї сполуки, яку вибирають з практичних міркувань.
Розрізняють середню та істинну (або миттєву) швидкості реакції.
Середня швидкість реакції ϑсер визначається різницею концентрацій [TEX]\Delta C[/TEX] речовини протягом певного часу [TEX]\Delta \tau[/TEX]:
- (1) [TEX]ϑ_{сер} = (C_2 – C_1)/(\tau_2 – \tau_1) = ± ΔC/ Δ\tau[/TEX],
- де С2 і С1 – концентрації речовини у кінцевий [TEX]\tau_2[/TEX] і початковий [TEX]\tau_1[/TEX] моменти часу.
Знак «±» у рівнянні (4.1) має такий зміст. Оскільки швидкість реакції завжди має додатне значення, то при використанні величини [TEX]\Delta C[/TEX] для вихідної речовини, концентрація якої протягом часу зменшується (С2,вих. < С1,вих, С2,вих. – С1,вих. = ΔCвих < 0), беруть знак мінус (рис. 4.3 а). Якщо швидкість визначають за змінюванням концентрації одного з продуктів реакції, кількість якого поступово зростає (С2,прод > C1,прод, С2,прод – С1,прод = ΔCпрод > 0), то відношення [TEX]\Delta C/\Delta \tau[/TEX] треба брати із знаком плюс (рис. 4.3 б).
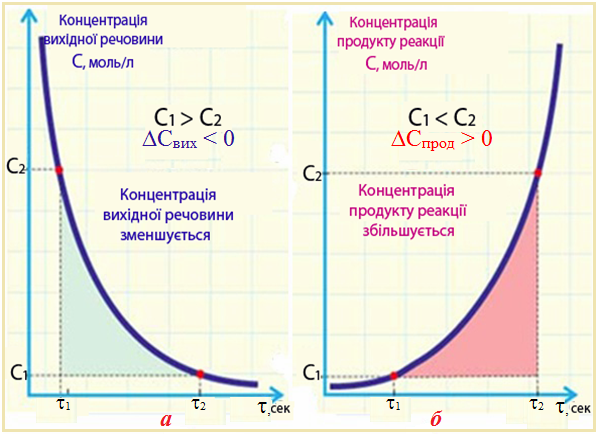
Рисунок 4.3 – Змінення концентрації протягом реакції:
а) вихідного реагенту; б) продукту реакції
У ході реакції змінюються концентрації реагуючих речовин і відповідно змінюється швидкість реакції. Чим коротший проміжок часу [TEX]\Delta \tau[/TEX], тим менше змінення концентрацій [TEX]\Delta C[/TEX] і тим ближче відношення [TEX]\Delta C/\Delta \tau[/TEX] до істинної (або миттєвої) швидкості реакції.
Однак концентрації речовин у хімічному процесі змінюються безперервно (рис. 4.4), тому правильніше говорити не про середню, а про істинну швидкість реакції, яка є похідною від концентрації за часом:
Як істинну, так і середню швидкості реакції можна визначити графічно – через тангенс кута нахилу дотичної (ϑіст) до кривої залежності концентрацій від часу чи через тангенс кута нахилу січної (ϑсер) (рис. 4.4):
ϑіст = tg α,
ϑсер = tgβ.
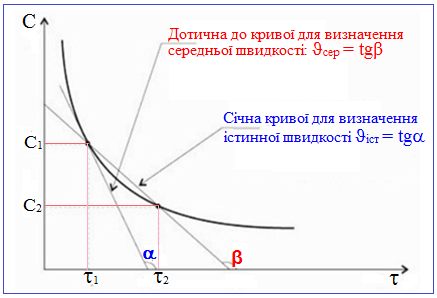
Рисунок 4.4 – Графічне визначення середньої швидкості (ϑсер = ±C / [TEX]\Delta \tau[/TEX] чи ϑсер = tgβ) та істинної швидкості реакції (ϑіст = tgα)
Із визначення швидкості реакції і аналізу рівняння (4.2) випливає, що швидкість реакції в одиницях СІ вимірюється у [TEX][моль\cdot м^{-3}\cdot с^{-1}][/TEX], однак використовуються й інші одиниці вимірювання [TEX][моль\cdot л^{-1}\cdot с^{-1}][/TEX], [TEX][моль\cdot см^{–3}\cdot с^{–1}][/TEX], [TEX][моль\cdot см^{–3}\cdot хв^{–1}][/TEX].
Упродовж реакції змінюються концентрації всіх реагентів і продуктів реакції. Для реакцій з різними стехіометричними коефіцієнтами швидкості змінення концентрацій реагентів теж будуть різними. Це необхідно враховувати при обчислюванні швидкості реакції. Так, для реакції загального вигляду
[TEX]aA+bB\rightarrow lL+mM[/TEX]
швидкість реакції визначається одним із співвідношень:
[TEX]\frac{\text{d}C_L}{l\text{d}\tau}=\frac{\text{d}C_M}{m\text{d}\tau}=-\frac{\text{d}C_A}{a\text{d}\tau}=-\frac{\text{d}C_B}{b\text{d}\tau}[/TEX].
На швидкість реакції впливають різні чинники (рис. 4.5), у першу чергу – природа реагуючих речовин: деякі реакції закінчуються миттєво, наприклад, при вибухах, а інші тривають роками (корозія). Крім того, швидкість реакції залежить від концентрації реагентів, розміру поверхні дотику фаз (для гетерогенних процесів), температури, каталізатора, зовнішніх чинників (наприклад, змінення тиску чи опромінювання).
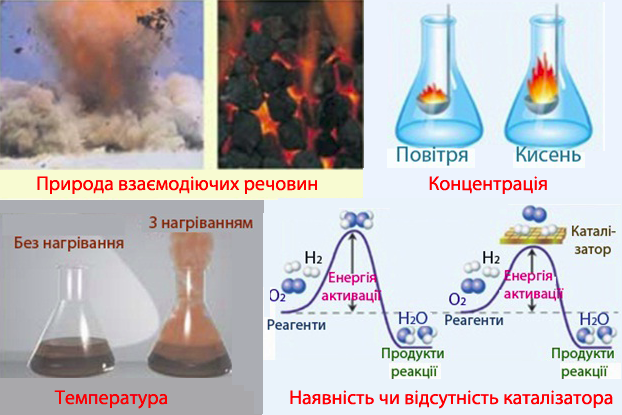
Рисунок 4.5 – Вплив різних чинників на швидкість реакції
Вплив природи речовин на швидкість реакції
Залежність швидкості реакції від концентрації реагентів
Будь-яка реакція може здійснюватися тільки за умов зіткнення молекул реагуючих речовин, тому швидкість реакції насамперед залежить від числа зіткнень, яке пропорційне концентрації реагентів. Ця закономірність була встановлена Гульдбергом і Вааге і одержала назву закон діючих мас, або закон Гульдберга-Вааге: швидкість хімічної реакції пропорційна добутку концентрацій реагуючих речовин у ступенях, які дорівнюють стехіометричним коефіцієнтам, що стоять перед формулами відповідних речовин у рівнянні реакції.
Для реакції між умовними речовинами A і B з утворенням продуктів L і M, (буквами [TEX]a,\;b,\;l[/TEX] і [TEX]m[/TEX] позначені коефіцієнти)
[TEX]aA+bB\rightarrow lL+mM[/TEX]
закон діючих мас має такий математичний вираз:
де CА і CВ – позначення концентрацій вихідних реагентів А і В, k – константа швидкості, яка не залежить від концентрації реагентів, але залежить від їх природи і температури.
Із рівняння (4.3) випливає фізичний зміст константи швидкості: при концентраціях вихідних реагентів СА = СВ = 1 моль/л (або за умови СА ⋅ СВ = 1) константа швидкості чисельно дорівнює швидкості реакції. При постійній температурі константа швидкості реакції між певними сполуками має сталу величину і характеризує природу реагуючих речовин.
Як довів досвід, закон Гульдберга-Вааге виявився справедливим тільки для обмеженого кола реакцій з невеликими стехіометричними коефіцієнтами, сума яких не перевищує 3, а для складніших процесів розрахунки за рівнянням (4.3) дають значну похибку. Це пов’язано з тим, що для більшості взаємодій сумарне рівняння реакції не відображає дійсного механізму процесу з безліччю проміжних стадій, а дає уяву тільки про склад і кількісне співвідношення вихідних речовин і продуктів реакції. Тому показники ступенів у рівнянні (4.3) не завжди повинні збігатися з стехіометричними коефіцієнтами. Насправді показники ступенів мають формальний характер і визначаються експериментально, а кінетичне рівняння відповідно до закону діючих мас набуває точнішого математичного вигляду:
де nA i nB – порядки реакції за відповідними реагентами, вони визначаються на практиці для кожної окремої реакції, а їх сума nА + nВ = n – це загальний порядок реакції, який і характеризує механізм процесу.
Порядок реакції за реагентом – це експериментально визначена величина, що дорівнює показнику ступеня, до якого необхідно піднести концентрацію даного реагенту, щоб теоретично розрахована швидкість реакції дорівнювала встановленій практично.
Поняття порядок реакції було введене в сучасне формулювання закону діючих мас: швидкість реакції пропорційна добутку концентрацій реагентів у ступенях, що дорівнюють порядкам реакцій за відповідними реагентами.
Реакції можуть мати різні порядки, у тому числі й дробові. Залежно від порядку реакції розглядають такі типи реакцій:
-
Реакції нульового порядку (рис. 4.6 а), що проходять з постійною швидкістю протягом часу (ϑ = const), яка не залежить від концентрацій реагуючих речовин. Нульовий порядок характерний для гетерогенних реакцій, для яких швидкість дифузії реагентів до поверхні поділу фаз менша, ніж швидкість їх безпосереднього хімічного перетворення. Наприклад, гідрування етилену на платиновому каталізаторі
[TEX]\rm C_2H_4+H_2 \xrightarrow[]{\;Pt-каталізатор\;}C_2H_6[/TEX],
для якого nА + nВ = 0. При цьому швидкість реакції не залежить від концентрації C2H4 і H2, а визначається лише зовнішніми умовами і поверхнею каталізатору. Тому кінетичне рівняння для цієї реакції має вигляд:
[TEX]\vartheta=k\cdot C_{C_2H_4}^0\cdot C_{H_2}^0[/TEX].
-
Реакції першого порядку залежать від концентрації тільки одного реагенту, тому описується кінетичним рівнянням
До таких реакцій належить дисоціація і розклад молекул, наприклад:
Н2→ 2H,
2N2O5(г)→ 4NO2(г) + O2,
CH3OCH3→ CH4 + H2 + CO.
Якщо прирівняти вирази (4.2) і (4.5) і розділити перемінні, одержимо
[TEX]kC=-\frac{\text{d}C}{\text{d}\tau}[/TEX], [TEX]\frac{\text{d}C}{C} =-k\text{d}\tau[/TEX]
Розв'язок цього рівняння при початкових умовах [TEX]C_{\tau=0}=C_0[/TEX] дає вираз:
Підставивши (4.5) у (4.6), знайдемо
Як видно, концентрація реагентів і швидкість реакції першого порядку зменшується за експоненціальним законом. Рівняння (4.7) можна записати у вигляді
звідки константа швидкості реакцій першого порядку дорівнює:
Вираз ln(C0/C) безрозмірний, тому константа швидкості реакції першого порядку вимірюється в [с–1]. За графіком залежності змінення концентрації реагенту з часом (рис. 4.6 б) легко визначити константу швидкості реакції, обчисливши tgα:
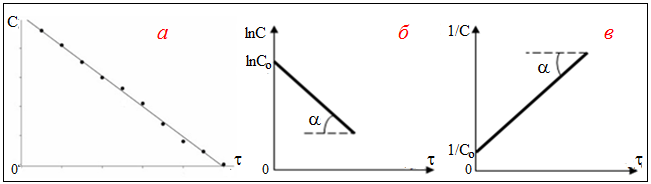
Рисунок 4.6 – Графік залежності концентрації реагенту від часу для реакції:
а) нульового порядку; б) першого порядку; б) другого порядку
-
Реакції другого порядку. Швидкість реакції другого порядку для умовних реагентів А і В підкоряється кінетичному рівнянню
ϑ = k CA·CB,
яке при однакових концентраціях вихідних реагентів (СА = СВ = С) набуває простішого вигляду:
Із зіставлення рівнянь (4.11) і (4.2: [TEX]\vartheta=\text{d}C/\text{d}\tau[/TEX]) одержуємо:
Розв′язок рівняння (4.12) для початкової умови [TEX]C_{\tau =0}=C_0[/TEX] дає:
звідки легко знайти константу швидкості реакції другого порядку:
Із аналізу одиниць вимірювання для виразу (4.14) встановлюється розмірність константи швидкості реакції другого порядку: [моль–1⋅л⋅с–1]. Константу швидкості можна визначити і графічно за експериментальною кривою (рис. 4.6 в)
Другий порядок мають, наприклад, такі реакції:
2HI → H2 + I2,
2NO2→ 2NO + O2.
Окислення бромоводню киснем за схемою
4HBr + O2 → 2Br2 + 2H2O
теж є реакцією другого порядку, незважаючи на те, що відповідно до рівняння реакції з однією молекулою кисню взаємодіють чотири молекули HBr. Розбіжність між коефіцієнтами в рівнянні реакції і математичним виразом для її швидкості
[TEX]\vartheta=kC_{\text{O}_2}\cdot C_{\text{HBr}}[/TEX]
пов’язана з тим, що у дійсності механізм реакції складається з трьох стадій:
- НВr + О2 → HО2Вr (лімітуюча, найповільніша стадія),
- НО2Вr + НBr → 2НОВr,
- 2НОBr + 2НВr → 2Н2О + 2Вr2,
__________________________________
4НВr + О2 → 2Н2О + 2Вr2.
Перша стадія у розглянутому механізмі – найповільніша, а друга і третя – дуже швидкі, тому вони практично не впливають на тривалість реакції. Швидкість реакції у цілому визначається найповільнішою стадією, яка називається лімітуюча стадія. Саме тому взаємодія між НВr і О2, швидкість якої однаково залежить від концентрацій обох реагентів, належить до реакцій другого порядку.
-
Реакції дробового порядку зустрічаються надзвичайно рідко. До них належить, наприклад, газофазний синтез фосгену з карбон (ІІ) оксиду і хлору:
СО + Cl2 → СОCl2.
Вираз для швидкості цієї реакції визначений експериментально і має вигляд:
ϑ = k [CO] · [Cl2]3/2.
Інший приклад реакції дробового порядку – розклад озону в присутності хлору:
[TEX]2\text{O}_3\xrightarrow[]{\;\;\;\text{Cl}_2\;\;\;}3\text{O}_2[/TEX].
Швидкість цієї реакції описується кінетичним рівнянням
ϑ = k [О3]3/2.
4.2.2 Енергія активації
Загальною умовою елементарного акту хімічної взаємодії є зіткнення частинок. Однак не кожне зіткнення реакційно здатних молекул завершується хімічною взаємодією між ними. Для того, щоб реакція дійсно відбулася, необхідна геометрична відповідність між активними центрами частинок, яка називається стеричний фактор (рис. 4.7). Інакше кажучи, для того, щоб взаємодія між реагентами розпочалася, необхідно відповідна просторова орієнтація частинок одна відносно одної. Найбільшого значення стеричний фактор набуває для великих за розміром і громіздких молекул, які особливо часто зустрічаються серед органічних сполук.
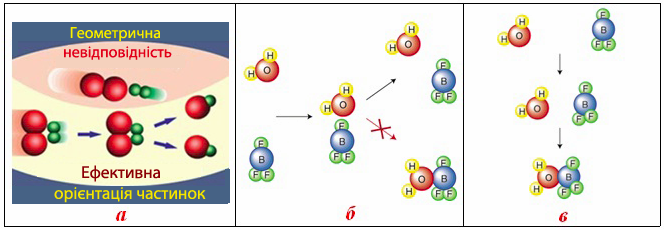
Рисунок 4.7 – Вплив стеричного фактору на хімічну взаємодію між молекулами: а) загальна схема дії стеричного фактору; б) реакція між молекулами Н2О і BF3 не відбувається, оскільки активні центри молекул не співпадають; в) взаємодія між Н2О і BF3 за умови відповідності активних центрів молекул, внаслідок чого утворюється продукт реакції BF3·Н2О
Під час хімічної реакції руйнуються одні хімічні зв’язки та утворюються інші, а це завжди супроводжується перерозподілом електронної густини, внаслідок чого частина молекул – так звані активні молекули – завжди має певний надлишок енергії порівняно з середньою енергією реакційної системи.
Активні молекули – це такі молекули, які внаслідок невпорядкованих зіткнень і перерозподілу енергії в системі набувають певного надлишку енергії та стають здатними до хімічної взаємодії.
Як доводить молекулярно-кінетична теорія газів і рідин, кількість зіткнень настільки велика, що всі реакції повинні відбуватися миттєво. Але цього не спостерігається і лише окремі зіткнення завершуються хімічною взаємодією, оскільки не всі частинки мають достатню енергію для подолання енергетичного бар’єру (рис. 4.8).
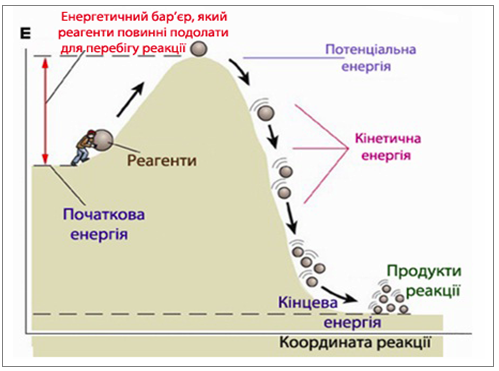
Рисунок 4.8 – Спрощена схема перебігу реакції через подолання енергетичного бар'єру
Виникнення енергетичного бар’єру зумовлюється необхідністю енергетичних витрат для розриву зв’язків у молекулах реагентів і відштовхуванням між їх електронними оболонками. Отже, не кожне зіткнення є ефективним (рис. 4.9).
Ефективні зіткнення – це такі, при яких енергія молекул є не тільки достатньою для розриву старих зв’язків у молекулах вихідних реагентів, але і перевищує енергію відштовхування (тобто енергетичний бар’єр) між електронними оболонками реагуючих частинок.
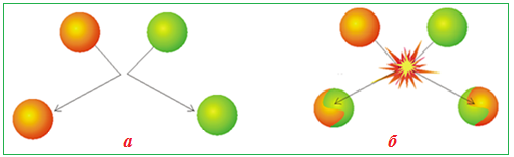
Рисунок 4.9 – Види зіткнень між частинками в реакційному просторі: а) неефективне зіткнення, при якому хімічна взаємодія не відбувається; б) ефективне зіткнення і утворення нових частинок
Під час ефективних зіткнень реакційна система проходить через проміжний стан, який називають активований комплекс – перехідний стан системи при хімічній реакції, коли старі зв’язки в молекулах вихідних реагентів ще не розірвані, але вже послаблені, а нові намітилися, але ще не утворилися. Наприклад, хід реакції, що описується рівнянням загального вигляду
![]()
можна виразити умовною схемою (рис. 4.10), в якій рисочками позначені існуючі хімічні зв'язки, точками – ті, що розриваються чи починають утворюватися, а буквами АВСD* – активований комплекс, у запису якого перелічують всі його складові частини і відмічають зірочкою.
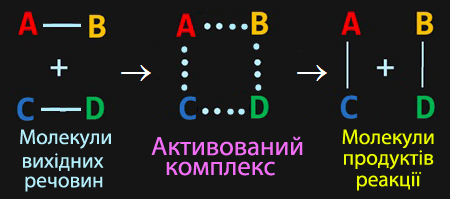
Рисунок 4.10 – Схема перебігу реакції АВ + СD → АС + ВD
через активований комплекс АВСD*
Час існування активованого комплексу дуже невеликий (приблизно 10–13 с). При його руйнуванні утворюються або продукти реакції, або знов вихідні речовини.
Енергія переходу речовини в стан активованого комплексу, яка дорівнює різниці між середньою енергією молекул реакційної системи і енергією, необхідною для перебігу хімічної реакції, називається енергія активації.
Під час хімічного процесу перехід системи від вихідних речовин з енергетичним станом Евих до продуктів реакції з енергетичним станом Епрод здійснюється через енергетичний бар’єр, який визначається енергією активації реакції Еакт. При цьому різниця енергій у вихідному і кінцевому станах дорівнює тепловому ефекту реакції:
ΔН = Епрод – Евих.
Графічно хід реакції зображується за допомогою енергетичної діаграми (рис. 4.11), в якій вісь ординат відображує енергію реакційної системи, а вісь абсцис – координату реакції, якою може бути будь-який контрольований параметр, що змінюється протягом реакції, наприклад: концентрація вихідної речовини чи продукту реакції, густина, об’єм газу, маса осаду тощо.
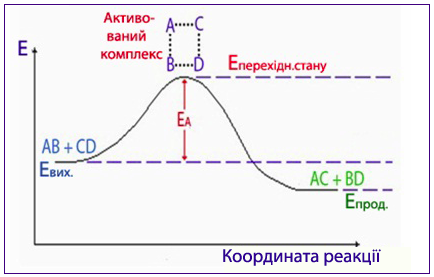
Рисунок 4.11 – Енергетична діаграма умовної реакції АВ + СD → АС + ВD, де ЕА – енергія активація, Евих. і Епрод. – середня енергія вихідних реагентів і продуктів реакції відповідно
Для прикладу розглянемо газофазну реакцію між двома оксидами нітрогену: N2O і NO (рис. 4.12), що проходить через активований комплекс [N2···O···NO*] згідно з рівнянням
N2O(г) + NO(г)↔ N2···O···NO* ↔ N2(г) + NO2(г).
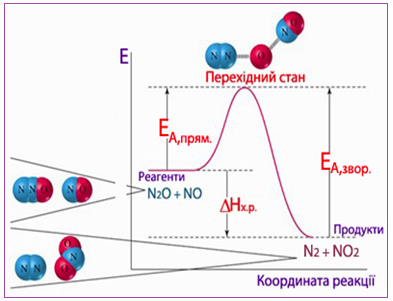
Рисунок 4.12 – Енергетична діаграма реакції між N2O і NO
З співставлення експериментально визначених величин енергії активації для прямої і зворотної реакцій (ЕА,прям = 209 кДж, ЕА,звор = 348 кДж) зрозуміло, що пряма реакція потребує меншої енергії активації, а зворотна – більшої. Крім того, з енергетичної діаграми видно, що пряма реакція є екзотермічною, оскільки енергія вихідних речовин вища, ніж енергія продуктів прямої реакції (Евих.прям > Епрод.прям). І навпаки, зворотна реакція – ендотермічна (Евих.звор < Епрод.звор). До такого висновку можна дійти, обчисливши теплові ефекти прямої та зворотної реакцій за різницею енергій активації:
ΔНпрям = ЕА,прям – ЕА,звор = 209 – 348 = –139 кДж,
ΔНзвор = ЕА,звор – ЕА,прям = 348 – 209 = +139 кДж
Оскільки ΔНпрям < 0, і ΔНзвор > 0, то зрозуміло, що пряма реакція є екзотермічною і супроводжується виділенням теплоти, а зворотна – ендотермічна, яка проходить з поглинанням теплоти. Слід звернути увагу, що енергія системи у перехідному стані активованого комплексу завжди має більшу величину, ніж у вихідному і кінцевому станах. Причому перебіг екзотермічних реакцій потребує меншої енергії активації, ніж ендотермічних (рис. 4.13).
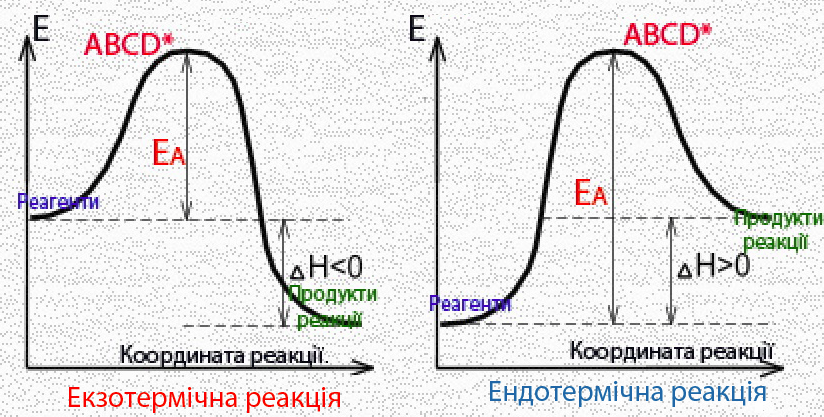
Рисунок 4.13 – Енергетична діаграма екзо- і ендотермічної реакції
Велика енергія активації (тобто високий енергетичний бар’єр) є причиною того, що багато хімічних реакцій за звичайних температур не відбуваються, незважаючи на їх принципову термодинамічну можливість (ΔG < 0). Так, за низьких температур самочинно не горять на повітрі нафта, вугілля, деревина, хоча для реакцій їх окислення енергія Гіббса ΔG має від’ємне значення.
4.2.3 Вплив температури на швидкість реакції
Підвищення температури зумовлює зростання загальної енергії реакційної системи, а це, в свою чергу, сприяє підвищенню швидкості руху і збільшенню відносного вмісту активних молекул. Вплив температури на швидкість реакції оцінюється за допомогою емпірично встановленої закономірності, яка називається правило Вант-Гоффа: підвищення температури на кожні 10 градусів збільшує швидкість реакції приблизно у 2-4 рази:
де (Т2 – Т1) = ΔТ – різниця температур між початковю Т1 і кінцевою Т2, ϑ1 і ϑ2 – початкова і кінцева швидкість реакції, [TEX]\gamma[/TEX] – температурний коефіцієнт швидкості, який показує, у скільки разів зростає швидкість реакції при підвищенні температури на десять градусів. Значення температурного коефіцієнта для ендотермічних реакцій вище, ніж для екзотермічних [TEX]\left(\gamma_{енд}>\gamma_{екз} \right)[/TEX]. Для більшості реакцій γ змінюється у межах 2-4.
Якщо перетворити рівняння Вант-Гоффа (4.15), поділивши ліву і праву його частини на ϑ1, одержимо рівняння вигляду
[TEX]\vartheta_2/\vartheta_1=\gamma^{(T_2-T_1)/10}[/TEX],
за допомогою якого легко обчислювати, у скільки разів швидкість реакції ϑ2 при кінцевій температурі Т2 більше (чи менше) початкової швидкості ϑ1 при температурі Т1.
Рівняння (4.15) зручно використовувати лише для приблизних розрахунків, тому що воно є справедливим за умови помірних температур і невеликого їх інтервалу. Точніше вплив температури на швидкість реакції відображає рівняння Арреніуса:
де k – константа швидкості реакції, k0 – передекспоненційний множник Арреніуса, пропорційний кількості зіткнень між молекулами, ЕА – енергія активації (для більшості хімічних реакцій ЕА = 40-400 кДж/моль).
Інтегрування рівняння Арреніуса (4.16) дає вираз для розрахунку енергії активації за відомими даними щодо констант швидкості реакції при двох температурах:
- (17) [TEX]\ln{\frac{k_{T_2}}{k_{T_1}}}=\frac{E_A}{R}\left( \frac{1}{T_1}-\frac{1}{T_2} \right)[/TEX]
або
- (18) [TEX]\ln{\frac{k_{T_2}}{k_{T_1}}}=\frac{E_A}{R}\left( \frac{T_2-T_1}{T_1\cdot T_2}\right)[/TEX].
Якщо концентрації реагуючих речовин дорівнюють 1 моль, то рівняння Арреніуса (4.16) дає змогу виразити залежність швидкості реакції від температури:
Згідно з рівнянням Арреніуса константа швидкості зменшується при зростанні енергії активації. Це рівняння дозволяє обчислювати константи швидкості (і саму швидкість) реакцій при різних температурах.
Вплив температури на швидкість реакції
4.3 Каталіз
Найбільш потужним засобом інтенсифікації хімічних процесів є застосування каталізаторів.
Каталізатор – це речовина, що збільшує швидкість реакції, кількісно і якісно при цьому не змінюючись. Явище змінювання швидкості реакції під впливом каталізатора називається каталіз.
Раніше каталізом називали будь-яке змінення швидкості реакції під впливом сторонніх речовин, які не витрачалися протягом реакції. Залежно від того, прискорюється чи сповільнюється швидкість, виділяли відповідно позитивний і негативний типи, причому негативні каталізатори називали інгібіторами. Такий поділ і дотепер можна зустріти у літературі, але останнім часом до нього звертаються все рідше.
Речовина, яка уповільнює швидкість хімічних процесів, а сама при цьому не змінюється, називається інгібітор.
Механізм дії інгібіторів є аналогічним дії каталізаторів – участь у проміжних стадіях процесу, наслідком чого є зниження числа активних молекул реагенту, які забезпечують перебіг реакції. Наприклад, атоми багатьох важких металів (Hg, Cd) в організмі людини реагують з молекулами білків, сповільнюючи життєво важливі біохімічні процеси.
Однак надалі зосередимося тільки на прискорюючому впливі каталізаторів.
Каталізаторам притаманні деякі специфічні особливості (рис. 4.14). Не піддаючись якісним і кількісним зміненням внаслідок реакції, каталізатори зменшують енергію активації, але не впливають при цьому на термодинамічні показники реакції (ΔН, ΔG, ΔS) і на константу хімічної рівноваги, рівною мірою збільшуючи швидкість як прямої, так і зворотної реакцій.

Рисунок 4.14 – Деякі специфічні особливості каталізаторів
Механізм дії каталізаторів дуже складний і не до кінця вивчений. Однак достовірно доведено, що вони зменшують енергію активації процесу, напрямляючи його перебіг іншим шляхом, через інші проміжні стани. Активований комплекс у присутності каталізатора має меншу енергію, ніж комплекс без каталізатора (рис. 4.15 а), тому енергія активації каталітичної реакції ЕА,к нижча за енергію активації некаталітичної реакції ЕА (рис. 4.15 б), тобто молекулам реагентів для взаємодії необхідно подолати значно нижчий енергетичний бар’єр.
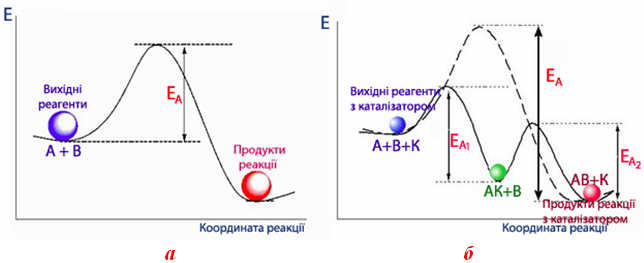
Рисунок 4.15 – Енергетична діаграма реакції: а) без каталізатора; б) за наявності каталізатора
За своїм агрегатним станом каталізатори бувають твердими, рідкими і газоподібними, тому каталітичні процеси поділяються на гомогенні і гетерогенні.
При гомогенному каталізі всі реагуючі речовини утворюють з каталізатором одну фазу (газоподібну або рідку). Механізм гомогенного каталізу пояснюється на основі проміжних сполук, які каталізатор утворює з реагентами і сприяє зменшенню енергії активації. Наприклад, реакція між умовними реагентами А і В, яка проходить через активований комплекс AB* за схемою
А + В → AB* → AB,
за наявності каталізатора перебігає через два (чи більше) проміжні стани:
А + Кat → AKat* → AKat,
AKat + B → AKatB* → AB + Kat.
До класичних прикладів гомогенного каталізу можна віднести реакцію окислення сульфур (IV) оксиду SO2(г) киснем O2(г) за участю каталізатора NO(г), що застовується у виробництві сульфатної кислоти (рис. 4.16):
[TEX]\rm 2SO_{2(г)}+O_{2(г)}\xrightarrow[]{\;\;\;NO_{(г)}\;\;\;}2SO_{3(г)}[/TEX]
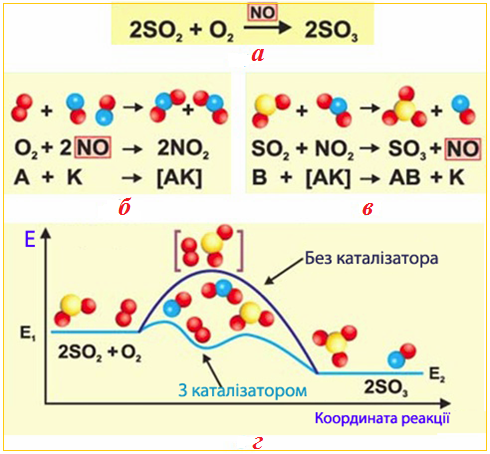
Рисунок 4.16 – Механізм гомогенного каталізу: а) загальне рівняння каталітичної реакції між реагентами SO2(г) і O2(г) за участю каталізатора NO(г); б) перша стадія, протягом якої каталізатор взаємодіє з однією з вихідних речовин – киснем, окислюючись до NO2; в) друга стадія, під час якої відбувається регенерація проміжної сполуки NO2 при її взаємодії з іншою вихідною речовиною (SO2) і відновлення початкової форми каталізатора (NO(г)); г) енергетична схема реакції 2SO2(г) + O2(г) → 2SO3(г) без каталізатора і за його наявністю
Часто при проведенні гомогенних реакцій використовуються такі газоподібні каталізатори, як атомарний хлор, пари йоду або водяна пара:
[TEX]\rm O_{3(г)}+O_{(г)}\xrightarrow[]{\;\;\;Cl_{(г)}\;\;\;}2O_{2(г)}[/TEX]
[TEX]\rm CH_3CHO_{(г)}\xrightarrow[]{\;\;\;I_{2(г)}\;\;\;}CH_{4(г)}+H_2O_{(г)}[/TEX]
[TEX]\rm 2CO_{(г)}+O_{2(г)}\xrightarrow[]{\;\;\;H_2O_{(г)}\;\;\;}2CO_{2(г)}[/TEX]
Більш поширеними є гомогенні каталітичні реакції в рідкій фазі, в яких роль каталізатора можуть виконувати розчинники (особливо вода), іони гідрогену або гідроксид-іони. Каталіз іонами Н+ і ОН- називають кислотно-основним. Прикладами гомогенного рідкофазного каталізу є такі процеси:
[TEX]\rm C_{12}H_{22}O_{11(р-н)}+H_2O\xrightarrow[]{\;\;\;H_2SO_{4(р)}\;\;\;}C_6H_{12}O_{6(р-н\;глюкози)}+C_6H_{12}O_{6(р-н\;фруктози)}[/TEX]
[TEX]\rm 2C_2H_5OH_{(р)}\xrightarrow[]{\;\;\;H_2SO_{4(р)}\;\;\;}C_2H_5-O-C_2H_{5(р)}+H_2O_{(р)}[/TEX]
[TEX]\rm CH_3CO-O-C_2H_{5(р)}+H_2O_{(р)}\xrightarrow[]{\;\;\;H_2SO_{4(р)}\;\;\;}CH_3COOH_{(р)}+C_2H_5OH_{(р)}[/TEX]
При гетерогенному каталізі реагенти і каталізатори перебувають у різних фазах і відокремлюються один від одного межею поділу. Як правило, гетерогенними є тверді каталізатори, на поверхні яких реагують газоподібні речовини. Сумарна швидкість перетворення на гетерогенному каталізаторі залежить від площі його поверхні (рис. 4.17), тому звичайно використовують каталізатори з розвиненою поверхнею або наносять їх тонким шаром на пористий носій (активоване вугілля, силікагель тощо).
Рисунок 4.17 – Моделі поверхонь гетерогенних каталізаторів
Існує декілька теорій гетерогенного каталізу. Згідно з найбільш вичерпною теорією Баландіна для здійснення каталізу необхідна геометрична відповідність між параметрами кристалічної решітки каталізатора і довжинами хімічних зв’язків у молекулах реагентів та продуктів реакції. У більшості теорій припускається, що реакція перебігає не на всій поверхні каталізатора, а лише на активних центрах – ділянках, де забезпечуються оптимальні умови процесу. Кількість активних центрів визначається складом поверхневого шару, способом приготування каталізатора і обробки його поверхні.
Реакція в присутності гетерогенного каталізатора проходить поетапно: спочатку на активних центрах поверхні послідовно адсорбуються реагенти, після чого між ними відбувається власно хімічна взаємодія і утворюється продукт. Наступним етапом є десорбція продукту з поверхні каталізатору і виведення його з реакційного середовища (рис. 4.18).
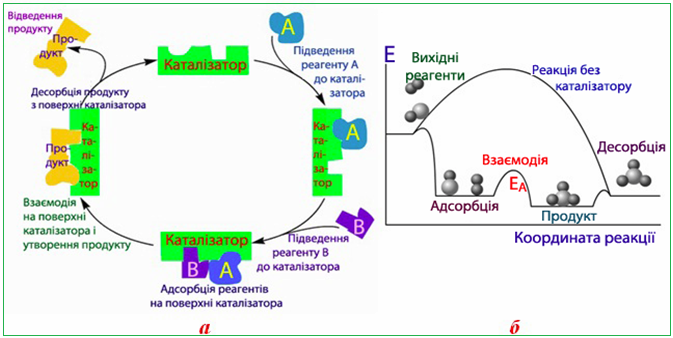
Рисунок 4.18 – Гетерогенний каталіз: а) механізм; б) енергетична діаграма
Типовим прикладом гетерогенного каталізу є процес окислення сульфур (IV) оксиду SO2(г) киснем O2(г) на поверхні твердого каталізатора V2O5(тв) (рис. 4.19).
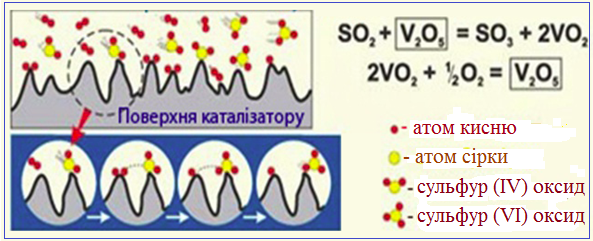
Рисунок 4.19 – Схема гетерогенного каталізу при одержанні сульфур (VІ) оксиду SO3 у сульфатнокислотному виробництві
В якості інших прикладів гетерогенного каталізу можна навести прискорення таких процесів: розклад гідроген пероксиду за участю манган (IV) оксиду
[TEX]\rm 2H_2O_{2(р)}\xrightarrow[]{\;\;\;MnO_{2(тв)}\;\;\;}2H_2O_{(р)}+O_{2(г)}[/TEX]
синтез амоніаку
[TEX]\rm N_{2(г)}+3H_{2(г)}\xrightarrow[]{\;\;\;Fe_{(тв)}\;\;\;}2NH_{3(г)}[/TEX]
виробництво метилового спирту
[TEX]\rm CO_{(г)}+2H_{2(г)}\xrightarrow[]{\;\;\;ZnO/Cr_2O_{3(тв)}\;\;\;}CH_3OH_{(р)}[/TEX]
гідрування етилену на платині (рис. 4.20)
[TEX]\rm C_2H_{4(г)}+H_{2(г)}\xrightarrow[]{\;\;\;Pt_{(тв)}\;\;\;}C_2H_{6(г)}[/TEX]
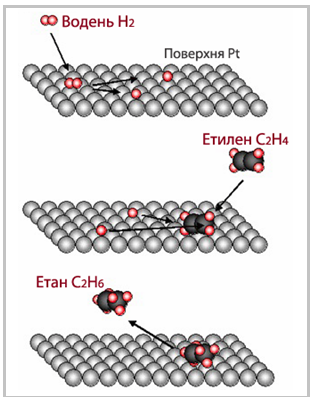
Рисунок 4.20 – Схема каталітичного гідрування етилену на платиновому каталізаторі
Важливою властивістю є селективність каталізаторів (вибірність) – здатність спрямовувати взаємодію одних і тих же самих речовин у різних напрямках для одержання бажаних продуктів.
Каталітична активність багатьох каталізаторів зростає при додаванні невеликих кількостей промоторів. Промотор – каталітично неактивна речовина, присутність якої посилює дію каталізаторів. Наприклад, швидкість окислення SO2 на каталізаторі V2O5 зростає в сотні разів при додаванні промоторів – сульфатів лужних металів.
У той же час існують речовини, які погіршують каталітичну активність каталізаторів – каталітичні отрути. Так, для платинових каталізаторів сильними каталітичними отрутами є сполуки Сульфуру, Арсену, Меркурію.
Каталізатори мають важливе значення, оскільки забезпечують економію енергії та сировини і допомагають вирішувати екологічні проблеми (очищення стічних вод, промислових та автомобільних викидів). Застосування каталізаторів стає необхідним і при створенні екологічно чистих маловідходних технологій.
Вплив каталізатора на швидкість реакції
Вплив інгібітора на швидкість реакції
4.4 Швидкість гетерогенних реакцій
Гетерогенні реакції відбуваються на поверхні поділу фаз, яка і вважається реакційним простором.
Швидкість гетерогенної реакції – це величина, яка визначається зміненням кількості речовини, що вступає в реакцію чи утворюється внаслідок неї за одиницю часу на одиниці площі поверхні фаз.
[TEX]\vartheta_{гетерог}=\pm \frac{\Delta \nu}{S\Delta \tau}[/TEX],
де [TEX]\Delta \nu[/TEX] – різниця між кількістю речовини ([TEX]\Delta \nu=\nu_2-\nu_1[/TEX] ) в кінцевий [TEX]\tau_2[/TEX] і початковий [TEX]\tau_1[/TEX] моменти часу, S – площа поверхні.
На практиці площу поверхні твердого тіла не завжди легко виміряти, тому іноді швидкість гетерогенної реакції відносять не до одиниці поверхні, а до одиниці маси чи до одиниці об’єму більш конденсованої фази.
Особливістю кінетики гетерогенних реакцій є вплив площі реакційної поверхні на швидкість реакції.
Залежність швидкості реакції від площі поверхні контакту фаз має дуже складний характер, але її якісне оцінювання є очевидним: при збільшенні площі дотику реагуючих речовин швидкість гетерогенної реакції зростає (рис. 4.21).
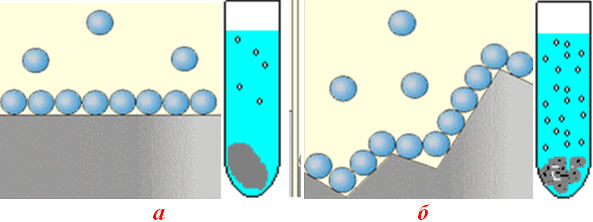
Рисунок 4.21 – Вплив площі дотику взаємодіючих фаз і стану поверхні на швидкість гетерогенної реакції
СаCО3(тв) + 2HCl(р-н) → CaCl2(р-н) + CO2↑(г) + H2O(г):
а) на ділянці, що має плоску поверхню, з твердою фазою (СаCО3) може одночасно контактувати менша кількість молекул HCl і, як наслідок, виділяється менше продуктів – бульбашок CO2; б) розвинена поверхня твердої фази забезпечує більше можливостей для контакту реагентів і утворення продуктів реакції
Вплив розміру поверхні дотику речовин на швидкість реакції
Слід пам’ятати важливе правило: якщо в гетерогенній реакції безпосередньо бере участь тверда речовина, то в кінетичне рівняння не входить його концентрація, яка вважається постійною протягом реакції. Наприклад, для гетерогенної реакції
СаО(тв) + СО2(г) → CaCO3(тв)
кінетичне рівняння має вигляд:
[TEX]\vartheta=k\cdot C_{\text{CO}_2}[/TEX].
Більшість гетерогенних реакцій складається з трьох основних стадій (рис. 4.22):
- підведення однієї реагуючої речовини до поверхні іншої;
- хімічна взаємодія на поверхні поділу фаз;
- відведення продукту від поверхні.
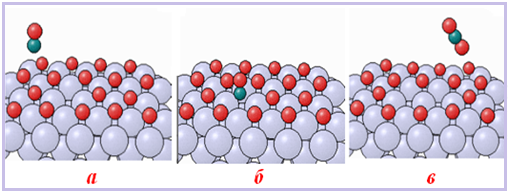
Рисунок 4.22 – Стадії гетерогенних реакцій: а) підведення однієї реагуючої речовини до поверхні іншої; б) хімічна взаємодія на поверхні; в) відведення продукту від поверхні
Найповільніша стадія, яка практично визначає швидкість реакції в цілому, називається лімітуюча стадія.
Якщо величина енергії активації хімічної реакції достатньо невелика, то лімітуючими стадіями є перенесення речовини. Для підвищення швидкості таких реакцій посилюють конвекцію – найчастіше за допомогою перемішування. Так, горіння вугілля, хімічна стадія якого потребує невеликої енергії активації, відбувається тим швидше, чим інтенсивніше подається до вугілля кисень.
Однак для реакцій з високою енергією активації лімітуючою є друга стадія, в цьому випадку переміщування не буде прискорювати взаємодію. Наприклад, іржавіння заліза на вологому повітрі не посилюється при збільшенні подачі кисню, оскільки енергія активації цієї реакції досить значна.
Другою особливістю швидкості гетерогенної реакції є її залежність від швидкості подачі реагенту в реакційну зону.
Найбільше змінення концентрації спостерігається у тонкому шарі реагенту поблизу реакційної поверхні, який називається дифузійний шар. Перенесення речовини в ньому здійснюється за рахунок дифузії. При перемішуванні товщина дифузійного шару зменшується і відповідно зростає швидкість підведення реагентів. Якщо швидкість дифузії нижча, ніж швидкість хімічної взаємодії, то лімітуючою стадією є дифузія. В такому випадку говорять, що має місце дифузійний контроль. Коли швидкість дифузії достатньо висока, то спостерігається кінетичний контроль, при якому процес лімітується власне хімічною реакцією. А якщо швидкості дифузії і хімічної реакції порівнянні, то має місце змішаний контроль.
Гетерогенні процеси мають важливе значення у техніці; до них належать корозія металів і сплавів, горіння твердого палива, випалювання сульфідних руд тощо.
4.5 Необоротні та оборотні хімічні реакції. Умова хімічної рівноваги
Внаслідок хімічної взаємодії між реагентами (вихідними сполуками) утворюються нові речовини – продукти реакції. В одних випадках на цьому взаємодія, яка називається прямою реакцією, завершується, а в інших – починається нова взаємодія, вже між утвореними продуктами, тобто відбувається зворотна реакція. Залежно від таких особливостей хімічної взаємодії реакції поділяються на необоротні та оборотні.
Необоротні реакції – це такі хімічні реакції, які перебігають лише у прямому напрямі і тривають до повного витрачання реагентів.
Умови необоротности хімічних реакцій були розглянуті в темі 2 (див. § 2.2). Зараз тільки згадаємо, що для необоротних реакцій притаманні такі ознаки: а) виділення осаду чи газу; b) утворення малодисоційованих сполук – слабких електролітів: води, слабкої кислоти чи слабкої основи; с) виділення великої кількості теплоти (горіння, вибух).
Приклади необоротних реакцій:
2KClO3 → 2KCl + 3O2↑,
Zn + 2HCl → ZnCl2 + H2↑,
2Al + 2KOH + 6H2O → 2K[Al(OH)4] + 3H2↑.
З точки зору термодинаміки, відповідно до рівняння Гіббса (ΔG = ΔH – TΔS) необоротні процеси супроводжуються зменшенням ентальпії (ΔН < 0) і збільшенням ентропії (ΔS > 0) – за таких умов енергія Гіббса завжди матиме від’ємне значення (ΔG < 0), а це свідчить про можливість самочинного протікання необоротних реакцій (див. § 3.8).
Оборотні реакції – це такі хімічні реакції, які можуть відбуватися як у прямому, так і в зворотному напрямах. Наприклад, реакція
2Н2 + О2↔ 2Н2О
при температурі 800-1500°С перебігає в прямому напрямку, а при 3000-4000°С – у зворотному. А для реакції
Н2 + І2↔ 2НI
при 300-400°С характерний перебіг як у прямому, так і зворотному напрямку одночасно (рис. 4.23).
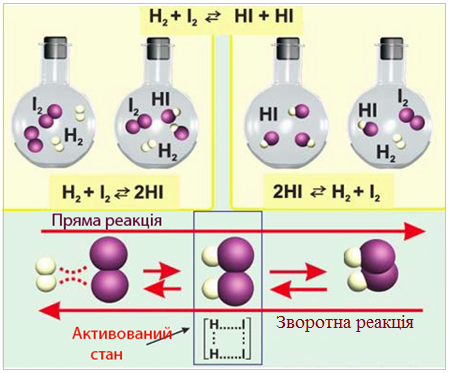
Рисунок 4.23 – Схема оборотної хімічної реакції
Отже, головною відмінністю оборотних реакцій є можливість перебігу прямої (→) і зворотної (←) реакцій. У рівняннях оборотних реакцій замість знаку рівності (=) або стрілки (→) використовують подвійну стрілку, напрямлену в протилежні боки (↔ чи ⇔).
Оборотні реакції найчастіше супроводжуються зменшенням ентальпії (ΔН < 0) і ентропії (ΔS < 0) системи, причому ΔG може мати від’ємне значення (якщо переважає ентальпійний фактор ΔН) або додатне (при високих температурах, коли переважає ентропійний фактор Т·ΔS). Для таких процесів за певних умов можливий перебіг як прямої, так і зворотної реакцій.
При деякій температурі ентальпійний (ΔН) і ентропійний (Т·ΔS) фактори можуть зрівнятися, дві протилежних тенденції будуть зрівноважувати одна одну, тобто ΔН = Т · ΔS і ΔG = 0. Це є термодинамічною умовою хімічної рівноваги.
Хімічна рівновага – це такий стан системи, при якому концентрації всіх речовин залишаються незмінними, а швидкості прямої та зворотної реакцій є однаковими.
Хімічна рівновага має динамічний характер. Це означає, що незмінність концентрації кожної речовини, що входить до складу реакційної системи, забезпечується не припиненням взаємодії, а тим, що швидкість прямої реакції дорівнює швидкості зворотної. З цієї причини кількість будь-якої речовини, що витрачається внаслідок перебігу однієї реакції, компенсується за рахунок утворення такої ж кількості цієї речовини у результаті реакції в протилежному напрямку (рис. 4.24).

Рисунок 4.24 – Оборотні реакції: до досягнення системою стану хімічної рівноваги швидкість прямої реакції зменшується (у міру витрачання вихідних речовин), а швидкість зворотної – навпаки, зростає завдяки збільшенню концентрації продуктів реакції. Після встановлення хімічної рівноваги швидкості прямої та зворотної реакцій зрівнюються
Подібний динамічний характер має хімічна рівновага і при фазових перетвореннях: випаровування ↔ конденсація; кристалізація речовини з розчину ↔ розчинення кристалів (рис. 4.25 а); сублімація парів з твердої фази ↔ кристалізація речовини з газоподібного стану (рис. 4.25 в), а також при хімічному процесі «димеризації молекул ↔ розпад димеру» (рис. 4.25 б), як це спостерігається при переході 2NO2 ↔ N2O4.
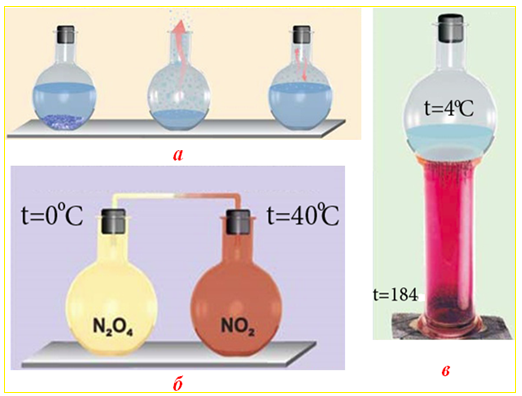
Рисунок 4.25 – Динамічний характер хімічної рівноваги при фазових і поліморфних переходах: а) кристали мідного купоросу з розчином CuSO4 (ліворуч) і з парами (праворуч); б) рівновага між молекулами NO2 і димерами N2O4; в) рівновага між твердим йодом I2(кр) і парами йоду I2(г)
4.6 Константа хімічної рівноваги
У стані хімічної рівноваги концентрації (або парціальні тиски газів у випадку газофазних взаємодій) вихідних речовин і продуктів реакції, називаються рівноважні концентрації (або рівноважними парціальними тисками).
Для реакції загального вигляду
[TEX]aA+bB\rightarrow lL+mM[/TEX]
швидкості прямої (ϑ1) і зворотної (ϑ2) реакцій відповідно до закону діючих мас виражаються кінетичними рівняннями:
[TEX]\vartheta_1=k_1[A]^a\cdot [B]^b[/TEX],
[TEX]\vartheta_2=k_2[L]^l\cdot [M]^m[/TEX],
де за допомогою квадратних дужок ([A], [B], [L], [M]) позначені рівноважні концентрації умовних речовин A, B, L і M, а буквами [TEX]a,\;b,\;l[/TEX] і [TEX]m[/TEX] – коефіцієнти перед формулами відповідних речовин. Нагадаємо, що при більш точних розрахунках замість коефіцієнтів використовують величини експериментально визначених порядків реакції за відповідними реагентами.
Відповідно до визначення, в стані хімічної рівноваги швидкості прямої та зворотної реакцій однакові (ϑ1 = ϑ2), тому можна прирівняти і праві частини кінетичних рівнянь:
[TEX]k_1[A]^a\cdot [B]^b=k_2[L]^l\cdot [M]^m[/TEX].
Якщо спочатку поділити ліву і праву частини одержаної рівності на константу швидкості зворотної реакції k2:
[TEX]\dfrac{k_1[A]^a\cdot [B]^b}{k_2}=\dfrac{k_2[L]^l\cdot [M]^m}{k_2}[/TEX]
а потім – на добуток концентрацій вихідних речовин [TEX][A]^a\cdot [B]^b[/TEX]:
[TEX]\dfrac{k_1[A]^a\cdot [B]^b}{k_2[A]^a\cdot [B]^b}=\dfrac{k_2[L]^l\cdot [M]^m}{k_2[A]^a\cdot [B]^b}[/TEX],
то після необхідних скорочень
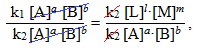
одержимо вираз
При даній температурі константи швидкості прямої k1 і зворотної k2 реакцій є величинами сталими, тому їх відношення k1/k2 теж стала величина, яка позначається великою літерою [TEX]K[/TEX] і називається константа рівноваги:
Аналогічний вигляд матиме константа рівноваги і для газофазної реакції [TEX]\left(aA_{(г)}+bB_{(г)}\leftrightarrow lL_{(г)}+mM_{(г)} \right)[/TEX], однак замість концентрації речовин використовують парціальні тиски газів [TEX]\left(P' \right)[/TEX]:
Рівняння (4.21) і (4.22) є варіантами математичного виразу закону діючих мас для стану рівноваги, який формулюється так:
При постійній температурі відношення добутку рівноважних концентрацій продуктів реакції до добутку рівноважних концентрацій вихідних речовин у ступенях, що дорівнюють стехіометричним коефіцієнтам (або точніше, порядкам реакції за відповідними реагентами), являє собою сталу величину і називається константа рівноваги.
Оскільки константа рівноваги пов’язана з енергією Гіббса рівнянням ізотерми Вант-Гоффа (див. § 3.8)
то за відомим значенням ΔG можна розрахувати константу хімічної рівноваги:
Якщо всі реагенти перебувають у газоподібному стані і підпорядковані законам ідеальних газів, то зв’язок між [TEX]K_c[/TEX] і [TEX]K_p[/TEX] можна виразити залежністю
де [TEX]\Delta \nu[/TEX] – змінення кількості речовини (моль) газів у результаті реакції: [TEX]\Delta \nu=(l+m)-(a+b)[/TEX].
Як випливає з рівнянь (4.24) і (4.25) константа рівноваги залежить від температури. Якщо в (4.23) підставити вираз енергії Гіббса (ΔG0 = ΔH0 – TΔS0) і провести перетворення, то одержимо:
[TEX]H^0-T\Delta S^0=-RT\ln{K_p}[/TEX],
[TEX]\ln{K_p}=-\dfrac{\Delta H^0}{RT}+\dfrac{\Delta S^0}{R}[/TEX],
або
Із зростанням абсолютного значення ΔН і зниженням температури чутливість константи рівноваги до змінювання температури підвищується.
4.7 Вплив зовнішніх чинників на хімічну рівновагу
Стан хімічної рівноваги за постійних умов може зберігатися будь-який час. Проте при зміненні умов стан рівноваги порушується.
Процес змінювання концентрацій речовин, викликаний порушенням стану рівноваги, називається зміщення хімічної рівноваги, або зсув хімічної рівноваги.
Змінення зовнішніх чинників може по-різному впливати на швидкість прямої та зворотної реакцій. Внаслідок цього хімічна рівновага зміщується в той чи інший бік. Якщо відбувається збільшення концентрацій речовин, що стоять у лівій частині рівняння реакції, то вважають, що рівновага зміщується вправо, тобто у напрямку прямої реакції. А при збільшенні концентрації речовин, що стоять у правій частині рівняння реакції, рівновага зміщується вліво, у напрямку зворотної реакції.
Через деякий час у системі знов встановиться рівновага, але вже за інших умов.
Характер зміщення рівноваги залежно від дії зовнішніх чинників визначає принцип Ле-Шательє (1882 р.): якщо на систему, що перебуває в стані хімічної рівноваги, подіяти зовнішнім чинником, то рівновага зміщується у напрямі процесу, який послаблює цю дію.
Принцип Ле-Шательє випливає з закону діючих мас. Якщо система за умов постійної температури перебуває у рівновазі, то при зовнішній дії константа рівноваги залишається сталою. Тому будь-яке змінення рівноважних концентрацій (або парціальних тисків газів) однієї чи декількох речовин приводить до такого змінення рівноважних концентрацій (парціальних тисків) інших речовин, яке забезпечує сталість константи рівноваги.
З принципу Ле-Шательє випливає ряд загальних положень.
- при збільшенні концентрації деякої речовини в рівноважній системі рівновага зміщується в бік витрачання цієї речовини; при зменшенні концентрації – у бік її утворення;
- при підвищенні температури рівновага системи, що перебуває у стані рівноваги, зміщується у напрямку ендотермічної реакції, а при зниженні – у бік екзотермічної;
- підвищення тиску приводить до зміщення рівноваги у бік утворення меншої кількості молекул газу (тобто речовин, які займають менший об′єм), а при зниженні тиску – у бік утворення більшої кількості молекул газу;
- якщо об′єм системи під час реакції не змінюється, то змінення тиску не впливає на стан рівноваги;
- каталізатор, однаково прискорюючи і пряму і зворотну реакції, не зміщує рівновагу, але сприяє її скорішому встановленню.
Надзвичайно наочним прикладом зміщення рівноваги під впливом зовнішніх чинників може бути рівноважна система
2NO2(г) ↔ N2O4(г) , ΔН < 0
завдяки різному забарвленню її складових частин: газ N2O4 безбарвний, NO2 – буро-оранжевий, а рівноважна суміш, що складається з NO2 і N2O4,– жовта (рис. 4.26 а).
З рівняння реакції видно, що пряма реакція – екзотермічна. Відповідно до принципу Ле-Шательє підвищення температури зміщує рівновагу у бік ендотермічної реакції (для даного прикладу – у бік зворотної), внаслідок якої збільшується кількість NO2, за рахунок чого колір набуває більш інтенсивного відтінку. І навпаки, охолодження рівноважної суміші прискорює перебіг прямої (екзотермічної) реакції – утворення більшої кількості безбарвного N2O4 (рис. 4.26 б).
Різна кількість молекул газу в обох частинах рівняння оборотної реакції свідчить і про вплив тиску: його підвищення зміщує рівновагу у бік прямої реакції (утворення безбарвного газу N2O4), яка дає меншу кількість молекул газу, тому забарвлення стає блідим. Зниження тиску зміщує рівновагу в системі (2NO2 ↔ N2O4) у бік зворотної реакції – утворення більшої кількості молекул газу (тобто у бік утворення бурого газу NO2), а колір стає темнішим (рис. 4.26 в).
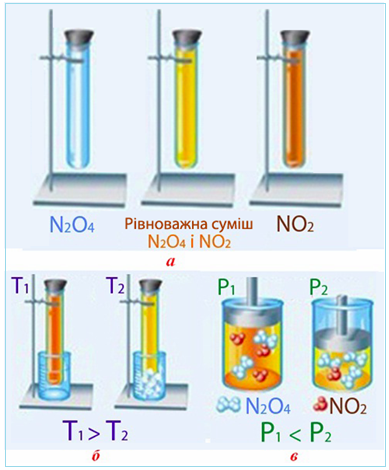
Рисунок 4.26 – Зміщення рівноваги в системі 2NO2 ↔ N2O4 + Q: а) забарвлення індивідуальних газів NO2, N2O4 та їх рівноважної суміші; б) вплив температури; в) вплив тиску
Принцип Ле-Шательє має велике практичне значення, особливо для хімічної промисловості. Однак він справедливий не тільки для хімічних реакцій, а поширюється на всі системи, що перебувають у стані динамічної рівноваги.
Хімічна рівновага
4.8 Приклади розв'язання типових задач
Приклад 4.1. Скласти кінетичне рівняння для гомогенної газофазної реакції
2H2S(г) + 3O2(г) → 2SO2(г) + 2H2O(г).
Розв′язок. Якщо відсутні дані про порядки реакції за відповідними реагентами, то у першому наближенні можна скористатися формулюванням закону діючих мас за Гульдбергом-Вааге і вважати, що швидкість реакції пропорційна добутку концентрацій вихідних речовин, піднесених у ступені, що дорівнюють стехіометричним коефіцієнтам перед формулами цих речовин у рівнянні реакції. Тоді для заданої реакції кінетичне рівняння має вигляд:
[TEX]\vartheta=k\cdot C_{\text{H}_2\text{S}}^2\cdot C_{\text{O}_2}^3[/TEX].
Приклад 4.2. Записати кінетичні рівняння для реакцій, що проходять за схемами:
а) СaCO3(тв) + 2HCl(р)→ CaCl2(р) + CO2(г) + H2O(р);
б) Fe2O3(тв) + 3H2(г)→ 2FeO(тв) + 3H2O(г).
Розв′язок. У випадку гетерогенних реакцій концентрація твердої фази вважається постійною і не вноситься в кінетичне рівняння, тому маємо:
а) [TEX]\vartheta=k_1\cdot C_{\text{HCl}(р)}^2[/TEX];
б) [TEX]\vartheta=k_1\cdot C_{\text{H}_2(г)}^3[/TEX].
Приклад 4.3. При 509° константа швидкості реакції Н2(г) + І2(г) → 2НI(г) дорівнює 0,16, а вихідні концентрації (моль/л): C(Н2)вих. = 0,04; C(І2)вих. = 0,05. Обчислити початкову швидкість реакції. Як зміниться швидкість реакції, коли концентрація водню зменшиться до 0,03 моль/л?
Розв′язок. Відповідно до закону діючих мас початкова швидкість реакції дорівнює:
ϑпоч = k ⋅ C(Н2)вих. ⋅ C(І2)вих. = 0,16 ⋅ 0,04 ⋅ 0,05 = 3,2 ⋅ 10–4.
До певного часу прореагувало водню: 0,04 – 0,03 = 0,01 моль/л.
Співставляючи коефіцієнти у рівнянні реакції, робимо висновок, що і йоду прореагувала така ж кількість, тому його концентрація набула значення:
C(І2) = 0,05 – 0,01 = 0,04 моль/л.
При цьому швидкість реакції
ϑ = k ⋅ C(Н2) ⋅ C(І2) = 0,16 ⋅ 0,03 ⋅ 0,04 = 1,92 ⋅ 10–4.
Порівняно із початковою швидкість реакції зменшиться у
ϑпоч / ϑ = 3,2 ⋅ 10-4 / 1,92 ⋅ 10-4 = 1,7 разів.
Приклад 4.4. Як зміниться швидкість газофазної реакції 2NO + O2 → 2NO2 при підвищенні тиску в 2 рази?
Розв′язок. Для спрощення позначимо вихідні концентрації реагентів: [NO]вих. = а, [O2]вих. = b . Тоді початкова швидкість реакції
ϑпоч = k ⋅ [NO]2 ⋅ [O2] = k ⋅ a2 ⋅ b.
При підвищенні тиску в 2 рази концентрації теж зростуть вдвічі:
[NO]1 = 2a; [O2]1 = 2b,
а швидкість реакції буде дорівнювати:
ϑ1 = k ⋅ (2a)2 ⋅ 2b = k ⋅ 4a2 ⋅ 2b = 8 ⋅ k ⋅ a2 ⋅ b = 8 ⋅ (k ⋅ a2 ⋅ b) = 8ϑпоч.
Тобто швидкість реакції зросте у 8 разів.
Приклад 4.5. У скільки разів зросте швидкість реакції 3А(г) + В(г) → 2D(г), що проходить у закритій посудині, якщо концентрації вихідних речовин збільшити у 3 рази?
Розв′язок. Позначимо концентрації (моль/л) вихідних речовин А і В буквами a і b відповідно і запишемо кінетичне рівняння:
ϑ = k ⋅ a3 ⋅ b.
Після збільшення концентрацій в 3 рази вони дорівнюватимуть 3a і 3b, тому швидкість реакції буде визначатися рівнянням
ϑ1 = k ⋅ (3a)3 ⋅ 3b,
а збільшення швидкості – відношенням ϑ1/ϑ. Тоді одержимо:
![]()
Таким чином, швидкість реакції зросла у 81 разів.
Приклад 4.6. Температурний коефіцієнт деякої реакції дорівнює 3. Як зміниться швидкість реакції при зниженні температури на 40°?
Розв′язок. Знайдемо змінення температури:
ΔТ = (Т2 – Т1) = –40.
Знак минус «–» підкреслює, що температура знижується. Перетворимо рівняння Вант-Гоффа (4.15) і підставимо дані:
![]()
Тобто при зниженні температури на 40° швидкість реакції зменшиться у 81 разів.
Приклад 4.7. При підвищенні температури від 40°С до 60°С швидкість реакції зросла у 9 разів. Чому дорівнює температурний коефіцієнт цієї реакції?
Розв′язок. Підвищення температури складає ΔТ = 60 – 40 = 20, тоді показник ступеню в рівнянні Вант-Гоффа дорівнює ΔТ / 10 = 20 / 10 = 2. Підставимо дані в перетворене рівняння Вант-Гоффа [TEX]\left(\vartheta_2/\vartheta_1=\gamma^{\Delta T/10} \right)[/TEX]:
ϑ2/ϑ1 = [TEX]\gamma[/TEX]2 = 9, звідки [TEX]\gamma[/TEX] = 3, оскільки 32 = 9.
Приклад 4.8. Записати вираз константи рівноваги для оборотних процесів:
а) N2(г) + 3H2(г) ↔ 2NH3(г);
б) 2Fe(OH)3(тв) + 6HCl(р) ↔ 2FeCl3(р) + 3H2O(р).
Розв′язок. а) Згідно з законом діючих мас для гомогенної системи у вираз константи рівноваги вносяться концентрації всіх речовин: ті, що у рівнянні реакції стоять зліва, наводяться у знаменнику, а ті, що справа, – у числівнику:
![]()
б) для гетерогенних реакцій концентрація більш конденсованої фази вважається постійною, тому її не зазначають у виразі константи рівноваги:
![]()
Одержані вирази констант рівноваги мають оцінювальний характер, оскільки для точнішого визначення константи рівноваги необхідно концентрації речовин підносити у ступені, що дорівнюють частинним порядкам реакції по кожному реагенту, а не тим коефіцієнтам, що стоять у рівнянні реакції перед формулами речовин.
Приклад 4.9. Обчислити константу рівноваги за деякої температури в газофазній системі 2NO2 ↔ 2NO + O2, якщо початкова концентрація NO2 дорівнювала 0,30 моль/л, а до встановлення рівноваги прореагувало 80 % NO2.
Розв′язок. Концентрація нітроген (IV) оксиду ([NO2]реакц), що вступив у реакцію та витратився внаслідок неї до моменту встановлення рівноваги (80%), складає:
[NO2]реакц = 0,30 · 0,80 = 0,24 моль/л.
Концентрація реагенту NO2, що не прореагував до встановлення рівноваги, і є його рівноважною концентрацією, яка дорівнює різниці між початковою концентрацією та тіією, що витратилася внаслідок реакції:
[NO2]рівн = [NO2]поч – [NO2]реакц = 0,30 – 0,24 = 0,06 моль/л.
З співставлення коефіцієнтів у рівнянні реакції (2NO2 ↔ 2NO + 1O2) видно, що до моменту рівноваги утворилося стільки NO, скільки розклалося NO2, і вдвічі менша кількість O2 порівняно з витраченою кількістю NO2, тобто:
[TEX]\nu[/TEX](NO)рівн = [TEX]\nu[/TEX](NO2)реакц,
[TEX]\nu[/TEX](O2)рівн = 1/2[TEX]\nu[/TEX](NO2)реакц.
Тоді рівноважні концентрації NO і O2 дорівнюють:
[NO]рівн = [NO2]реакц = 0,24 моль/л,
[O2]рівн = 1/2[NO2]реакц = 1/2·0,24 = 0,12 моль/л.
Підставляємо рівноважні концентрації всіх речовин у вираз константи рівноваги:
![]()
Приклад 4.10. Рівновага в системі 2Cl2(г) + 2H2O(г) ↔ 4HCl(г) + O2(г) встановилася при таких концентраціях (моль/л): [Cl2] = 0,8; [H2O] = 2,4; [HCl] = 1,2; [O2] = 1,4. Обчислити константу рівноваги і вихідні концентрації Cl2 і H2O, вважаючи, що на початку реакції хлороводень у системі був відсутній.
Розв′язок. Згідно із законом діючих мас (4.21) константа рівноваги дорівнює:
![]()
Позначимо концентрацію витраченого Cl2 через х і складемо пропорцію з урахуванням коефіцієнтів у рівнянні реакції
2Cl2(г) + 2H2O(г) ↔ 4HCl(г) + 1O2(г)
2 моль Cl2 –––––––––––––––––→ 4 моль HCl,
x моль/л Cl2 –––––––––––––––→1,2 моль/л HCl,
![]()
Оскільки до встановлення рівноваги витрачено 0,6 моль/л Cl2, то його вихідна концентрація дорівнювала:
[Cl2]вих = [Cl2] + x = 0,8 + 0,6 = 1,4 моль/л.
Перед формулою Н2О в рівнянні реакції стоїть такий самий коефіцієнт, що й перед формулою Cl2, звідки робимо висновок, що витрачена концентрація Н2О теж дорівнює 0,6 моль/л. Тоді вихідна концентрація водяної пари:
[H2O]вих = [H2O] + x = 2,4 + 0,6 = 3,0 моль/л.
Приклад 4.11. Обчислити рівноважні концентрації речовин у газофазній системі СО(г) + Н2О(г) ↔ СО2(г) + Н2(г) при 1023 К, якщо константа рівноваги Кс = 1, а початкові концентрації вихідних сполук [СО]поч = 3 моль/л; [Н2О]поч = 3 моль/л.
Розв′язок. Оскільки початкові концентрації продуктів не вказані, то вважаємо, що вони дорівнювали нулю: [CO2]поч. = 0; [H2O]поч = 0. Припустимо, що концентрація СО2 у ході реакції збільшилася на х моль/л, тоді рівноважна його концентрація дорівнює:
[CO2] = 0 + x = x.
З рівняння реакції видно, що на стільки ж збільшилася концентрація водню
[Н2] = 0+х = х
і зменшилися концентрації вихідних речовин, тобто
[CO] = 3–x і [Н2О ] = 3–х.
Підставимо рівноважні концентрації у рівняння закону діючих мас (8.2):
![]()
Звідки одержуємо:
9 – 6х + х2 = х2,
6х = 9,
х = 1,5.
Тепер можна обчислити рівноважні концентрації всіх речовин:
[CO] = 1,5 моль/л;
[H2] = 1,5 моль/л;
[H2O] = 3 – x = 1,5 моль/л;
[CO2] = 3 – x = 1,5 моль/л.
У тому випадку, коли константа рівноваги невідома, її можна обчислити на основі термодинамічних даних реакції, тобто:
[TEX]K_p=\exp (-\Delta G^0/RT)[/TEX] і [TEX]K_c=K_p(RT)^{-\Delta \nu}[/TEX].
Приклад 4.12. Як впливає підвищення тиску на стан рівноваги в оборотній гетерогенній системі Fe3O4(тв) + CO(г) ↔ 3FeO(тв) + CO2(г)?
Розв′язок. Запишемо вирази швидкостей прямої (ϑ1) та зворотної (ϑ2) реакцій:
ϑ1 = k1 [CO]; ϑ2 = k2 [CO2].
Швидкість гетерогенної реакції не залежить від концентрації твердої фази (Fe3O4(тв) і FeO(тв)), а число молекул газу в лівій і правій частинах рівняння реакції є однаковим, тому змінення тиску однаковою мірою змінює швидкість як прямої, так і зворотної реакцій і зміщення рівноваги не відбувається.
Приклад 4.13. В якому напрямку буде зміщуватися рівновага в гомогенній газофазній системі 2SO2 + O2 ↔ SO3, якщо при постійній температурі збільшити тиск у 4 рази?
Розв′язок. Відповідно до закону діючих мас швидкості прямої (ϑпр) і зворотної (ϑзвор) реакцій виражаються за допомогою кінетичних рівнянь:
ϑпр = k1·[SO2]2·[O2],
ϑзвор = k2·[SO3]2.
Збільшення тиску в 4 рази викличе збільшення концентрації кожної речовини у системі теж у 4 рази. Тоді швидкості прямої (ϑ'пр) і зворотної (ϑ'звор) реакцій зростуть так:
ϑ'пр = k1·(4[SO2])2·4[O2] = 64k1·[SO2]2·[O2] = 64ϑпр,
ϑ'звор = k2·(4[SO3]) 2= 16k2·[SO3]2= 16ϑзвор.
Розрахунки доводять, що при підвищенні тиску в реакційній системі в 4 рази швидкість прямої реакції зросла в 64 рази, а швидкість зворотної – тільки у 16 разів. З цього випливає, що хімічна рівновага зміщується у бік перебігу прямої реакції.
Приклад 4.14. Як буде зміщуватися рівновага в системі СН4 + 2Н2О(г) ↔ СО2 + 4Н2, ΔН > 0 :
а) при додаванні СН4;
б) при збільшенні тиску;
в) при підвищенні температури?
Розв′язок. а) При додаванні в рівноважну систему реагенту СН4 його концентрація збільшується, а це, відповідно до принципу Ле-Шательє, прискорює пряму реакцію, внаслідок якої відбувається витрачання доданої речовини. Тому рівновага буде зміщуватися вправо, у бік прямої реакції. Прискорення прямої реакції приведе до збільшення концентрації продуктів реакції СО2 і Н2 і, одночасно, до зменшення концентрації водяної пари Н2О(г). Процес буде проходити до тих пір, поки не встановиться нова рівновага з такими концентраціями всіх компонентів, щоб співвідношення між ними, яке визначається константою рівноваги, залишалося постійним.
б) Згідно з наслідками принципу Ле Шательє збільшення загального тиску в рівноважній системі зміщує рівновагу в бік зменшення тиску, що реалізується при зменшенні кількості молекул газу. А це досягається при протіканні зворотної реакції, оскільки у рівнянні реакції сумарна кількість речовини газу зліва (∑[TEX]\nu[/TEX] = 3 моль) більша, ніж справа (∑[TEX]\nu[/TEX] = 5 моль).
в) Оскільки пряма реакція ендотермічна (ΔН > 0), то підвищення температури у системі буде зміщувати рівновагу вправо – саме у бік ендотермічної реакції. Це можна поясними таким чином. Підвищення температури спричиняє збільшення константи рівноваги:
![]()
тому необхідно, щоб добуток концентрацій продуктів реакції зростав значніше, ніж добуток концентрацій вихідних речовин, що реалізується лише при зміщенні рівноваги у бік прямої реакції.
Приклад 4.15. При деякій температурі у газофазній системі А(г) + В(г) ↔ С(г), ΔН0 < 0 встановилася рівновага. Як впливає на концентрацію речовини С: а) підвищення тиску; б) зменшення концентрації речовини А; в) зростання температури; г) додавання каталізатору?
Розв′язок. а) З принципу Ле-Шательє випливає, що підвищення тиску зміщує хімічну рівновагу в бік утворення меншої кількості молекул газу, в даному випадку – в бік прямої реакції, внаслідок якої утворюється речовина С. Отже, концентрація речовини С збільшиться. До того ж висновку можна прийти на підставі простих розрахунків згідно з законом діючих мас. Позначимо рівноважні концентрації речовин:
[А] = a, [В] = b, [С] = c.
Тоді швидкості прямої ϑ1 і зворотної ϑ2 реакцій у стані рівноваги дорівнюють:
ϑ1 = k1a·b,
ϑ2 = k2c.
Припустимо, що тиск у системі підвищився у n разів. Відповідно до цього концентрація кожної речовини теж зросла у n разів, а швидкості прямої (ϑ'пр) і зворотної (ϑ'звор) реакцій тепер стали такими:
ϑ'пр = k1na·nb = n2k1a⋅b = n2ϑ1,
ϑ'звор = k2n·c = nk1c = nϑ2.
Видно, що при підвищенні тиску в n разів швидкість прямої реакції зросла значніше (у n2 разів), ніж швидкість зворотної реакції (тільки у n разів), тому і рівновага зміщується у бік прямої реакції, що приведе до збільшення концентрації речовини С.
б) Зменшення концентрації вихідної речовини А відповідно до принципу Ле-Шательє буде сприяти зміщенню рівноваги у бік реакції, внаслідок якої відбувається утворення цієї речовини, тобто у бік зворотної реакції. А це приведе до витрачання речовини С і, отже, до зменшення його концентрації.
в) Оскільки пряма реакція екзотермічна (ΔН0 < 0), то теплота виділяється. Тоді згідно з першим законом термохімії (Лавуазьє-Лапласа) зворотна реакція – ендотермічна. А з принципу Ле-Шательє випливає, що підвищення температури зміщує рівновагу у бік ендотермічної реакції (у даному випадку – зворотної), яка проходить з поглинанням теплоти. Тому концентрація речовини С зменшиться.
г) Додавання каталізатору рівною мірою прискорює пряму і зворотну реакцію, не зміщуючи рівновагу, тому відносна концентрація речовини не зміниться.

